2020年EMA和PMDA率先批准上市的18个新药逐个评述
2021-01-01
免疫疗法ASCO会议抗体创新药放射疗法
2020年是不平凡的一年,尽管疫情严重,但全球仍有70余个全新的药物获批上市。虽然EMA和PMDA批准的产品数量不及FDA,但以下18个率先批准的产品均别具特色,最值得关注的当属无活体供体的高敏器官移植用药,硼中子疗法和光免疫疗法。笔者在此对2020年获得EMA和PMDA批准的18新药一一简单介绍,以为业内同仁提供初步的了解。文章经过校对,每一个数据都有参考文献可追溯,如有疑问可联系作者Voyager88,本文不允许其他媒体转载,严禁抄袭、摘取文章内容发表至其它平台,侵权必究。
欧洲首次批准的新药
欧洲是全球第二大创新药市场,但欧洲国家众多,法规体系较为复杂,而且EMA的审速度不及FDA,故新药申请和审批的节奏相较美国慢。EMA在2020年批准的众多新药中,仅有5个产品为全球首次批准,其中3个是尚未获FDA批准的产品,其中最值得关注的产品为Idefirix,该产品的上市将为高致敏性的非活体器官移植提供可能。对于已经获得FDA批准的产品,本文将以FDA公开的资料为重点进行介绍。
1. Isturisa(osilodrostat)
1月9日,EMA首次批准了Recordati公司的osilodrostat,该产品于3月6日获得FDA批准,用于不适宜垂体手术或经过手术未治愈的成年库欣病患者治疗。库欣病是由垂体瘤引起,垂体释放出过多促肾上腺皮质激素引起肾上腺皮质醇合成过量,进而引发一些系列的临床症状。库欣病是一种极为罕见的疾病,患病率仅为4/10万,年发病率仅为0.7/100万-2.4/100万[79]。Osilodrostat是一种皮质醇合成抑制剂,可抑制11β-羟化酶的活性,阻断肾上腺合成皮质醇。FDA批准本品上市是基于一项有137名患者参与的临床试验(NCT02180217)的结果,经过24周的开放标签治疗,约有一半患者的皮质激素水平下降至正常范围内。在此基础上,71名无需增加治疗剂量(30mg每日2次)且在12周内耐受该药物的患者被纳入为期8周的双盲、随机停药研究,结果显示,继续使用osilodrostat治疗的患者中,86%的患者皮质激素维持在正常水平,而停药改用安慰剂的患者仅为30%[80,81]。市场潜力方面,EVP预测本品在2024年的销售额为4000万美元。
7月31日,EMA批准了MYR GmbH公司的bulevirtide,用于血浆/血清丁肝病毒(HDV)RNA呈阳性的慢性代偿性丁型肝炎RNA呈阳性的慢性代偿性丁型肝炎治疗。Bulevirtide是一种脂肪酸酰化的化学合成多肽,是一种钠离子-胆汁酸协同转运抑制剂,可阻止病毒进入肝细胞。在一项代号为MYR-202的临床研究中,118名慢性丁肝患者在替诺福韦的背景治疗下,添加不同剂量的bulevirtide或安慰剂,持续治疗24周,结果显示本品2mg治疗组有53.6%的患者,体内无法检测到HDV 病毒的RNA或RNA水平下降2个log10值以上,5mg组为50.0%,10mg组为76.7%,而安慰剂组仅为3.6%。除此以外,另一项代号为MYR-203的临床试验初步证明了产品长期用药的安全有效性,15名患者使用本品2mg治疗48周,疗效和安全性情况相比24周无显著性差异[82]。丁型病毒性肝炎是由丁型肝炎病毒(HDV)与乙型肝炎病毒等嗜肝DNA病毒共同引起的传染病,相比单纯的HBV感染者,HDV/HBV共感染者更容易转变为肝硬化或肝癌[83]。丁肝患者非常罕见,卫生健康委发布的数据显示,我国年发病人数为300-400人[84],为了扩大用药人群,bulevirtide还在开展乙肝方面的研究。
N -Tetradecanoylglycyl-Thr-Asn-Leu-Ser-Val-Pro-Asn-Pro-Leu-Gly-Phe-Phe-Pro-Asp-His-Gln-Leu-Asp-Pro-Ala-Phe-Gly-Ala-Asn-Ser-Asn-Asn-Pro-Asp-Trp-Asp-Phe-Asn-Pro-Asn-Lys-Asp-His-Trp-Pro-Glu-Ala-Asn-Lys-Val-Gly-NH2
7月31日,EMA批准了吉利德的filgotinib,适应症为单用或与甲氨蝶呤联用,治疗一种或多种疾病缓解性抗风湿药(DMARD)响应不足或不耐受的中重度活动性类风湿关节炎(RA)患者。Filgotinib是一种选择性JAK1抑制剂,相比非选择性JAK抑制剂,本品具有更好的安全性[85]。EMA批准本品上市,主要是基于三项3期临床试验(FINCH 1、2和3)的结果。在FINCH 1中,1755名甲氨蝶呤响应不足的RA患者,在甲氨蝶呤背景治疗的基础上分别给予本品200mg、100mg、阿达木单抗或安慰剂治疗,结果显示,四个治疗组在第12周的ACR20(主要终点,ARC20为美国风湿病学会指定的一种疗效评价标准)分别为77%、70%、71%和50%。FINCH 2(NCT02873936)是针对DMARD响应不足的RA患者设计的临床试验,448名患者在DMARD的背景治疗下分别添加本品200mg、100mg或安慰剂治疗,结果显示,三个治疗组在第12周的ACR20(主要终点)分别为66%、58%和31% [86]。FINCH 3是针对甲氨蝶呤初治患者设计的临床试验,1249名患者分别本品200mg+甲氨蝶呤、本品100mg+甲氨蝶呤、本品200mg或甲氨蝶呤单药治疗,四个治疗组在第24周的ACR20(主要终点)分别为81%、80%、78%和71%,除本品200mg单药剂量组外,其余两组均与甲氨蝶呤单药组产生显著性差异。不良反应主要是恶心、上呼吸道感染、尿路感染和头晕[87]。除了RA,本品还在开发溃疡性结肠炎、克罗恩病、银屑病关节炎、强直性脊柱炎等多种免疫性疾病,EVP预测该产品在2026年的销售额可达14.04亿美元。
Preview
来源: 药事纵横
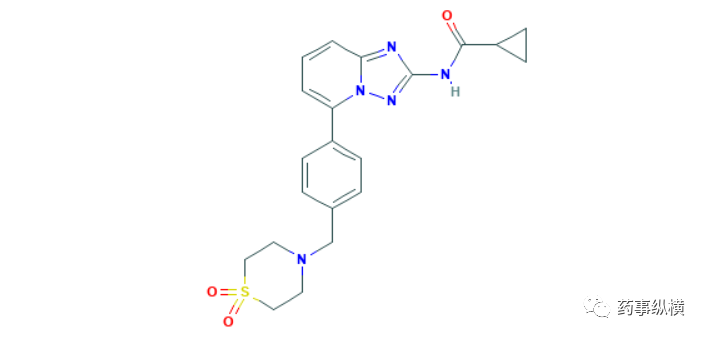
8月25日,EMA有条件批准了Hansa Biopharma公司的imlifidase,用于与已故捐赠者交叉匹配显阳性、高敏成人肾移植患者的脱敏治疗。对于供者特异性抗体(DSA)较高的患者,极易发生抗体介导的排斥反应(AMR)。脱敏就是为了安全移植而去除DSA的过程,对于有活体供体,可以预先置换血浆或注射免疫球蛋白实现脱敏,但对于没有活体供体而言,脱敏过程是器官移植的一大障碍。Imlifidase是一种化脓性链球菌的内肽酶,对人IgG(免疫球蛋白)具有特异性,可迅速清除IgG,提高移植的成功率[88]。EMA批准imlifidase是基于4项临床试验结果,其中试验一是一项开放标签的临床试验,46名肾移植患者(41名为高敏),交叉匹配阳性的患者在术前24小时内输注imlifidase,药动-药效学模拟显示,imlifidase输注后6个小时,99.5%的患者匹配测试转化为阴性。46名患者在移植后6个月全部存活,90%患者的肾功能恢复到正常水平。试验三与试验一的设计类似,10名肾移植患者(6名为交叉匹配阳性),使用imlifidase治疗后,全部患者移植成功,其中8名患者在移植后6个月时的肾功能恢复到正常水平。试验四评估了imlifidase在HLA-高敏患者中的安全有效性,17名患者(14名为交叉匹配阳性)接受imlifidase治疗后,16名在移植后第六个月的肾功能恢复到正常水平,虽然两名患者出现了AMR,但未导致移植失败。试验六与的设计与前几项试验类似,18名患者接受imlifidase治疗后,全部移植成功,16名患者肾功能恢复到正常水平[89]。Imlifidase的获批是肾移植领域的一大重要进展,除了肾移植,该产品开发的适应症还包括抗肾小球基底膜病、Guillain-Barre综合征和血栓性血小板减少症等。
11月19日,EMA首次批准了Alnylam公司的RNAi药物lumasiran,3天之后,该产品又获得FDA批准,用于1型原发性高草酸尿症(PH1)治疗。原发性高草酸尿症(PHs)是由编码丙氨酸-乙醛酸氨基转移酶的基因突变所引起的罕见遗传病,在西方国家的患病率为十五万分之一。基因缺陷所致的丙氨酸-乙醛酸氨基转移酶缺乏,可引起草酸生成异常升高,而Lumasiran是一种干扰RNA,可沉默编码HAO1(乙醇酸氧化酶)的mRNA,HAO1缺失可导致从乙醇酸到乙醛酸的转化过程受阻,进而影响草酸的合成[90]。FDA批准lumasiran上市,是基于两项临床试验的结果。试验一是一项针对6岁以上年龄段人群开展的,随机、双盲、安慰剂对照的临床试验,主要终点为24小时内尿液中的总草酸含量变化。结果显示,26名接受lumasiran治疗的患者,平均尿液中总草酸含量相比基线下降了65%,而13名接受安慰剂治疗的患者仅平均下降了13%。经过6个月的治疗,62%的lumasiran治疗患者24小时总尿酸含量达正常水平,而安慰剂组无一患者达到正常水平。试验二是一项开放标签的临床试验,主要针对6岁以下儿童设计,16名患者经过lumasiran为期6个月的治疗后,平均尿液总草酸含量相比基线下降了71%[91]。Lumasiran是首个获批用于该疾病治疗的药物,EVP预测该产品在2026年的销售额为2.53亿美元。
日本首次批准的新药
硼中子疗法和光免疫疗法均
1月23日,PMDA批准了日本烟草(Japan Tobacco)的delgocitinib软膏,用于特应性皮炎治疗。Delgocitinib是一种JAK(Janus kinase)抑制剂,PMDA批准该产品是基于一项三期临床试验(QBA4-1)的结果,156名中重度特应性皮炎患者,接受本品或安慰剂持续治疗4周,主要终点为第4周的湿疹面积及严重程度指数(EASI评分)相比基线的改善程度。结果显示,本品治疗组的EASI评分相比基线下降了44.3%,而安慰剂组却升高了1.7% [92]。另一项针对儿童患者开展的2期临床试验(JapicCTI-173553)结果同样证明了本品的有效性,受试者分别接受本品0.25%、0.5%两个不同含量的软膏或安慰剂治疗,治疗结果显示,三组患者的EASI评分相比基线下降54.2%、61.8%和4.8%。特应性皮炎是一种瘙痒性湿疹性皮炎,流行病学数据显示,该病在成人中的患病率为2%-10%,儿童更是高达10%-35%,具有非常庞大的治疗需求[93]。除了特应性皮炎,该产品还在开发斑秃、红斑狼疮等多种自身免疫性疾病。
Preview
来源: 药事纵横
2. Urece(dotinurad)
1月23日,PMDA还批准了富士药品(Fuji Yakuhin)的dotinurad,用于痛风和高尿酸血症治疗。Dotinurad是一种新型选择性尿酸盐转运蛋白抑制剂,PMDA批准该产品上市是基于在日本开展的三项3期临床试验的结果。试验一(NCT03100318)是一项随机、双盲的非劣性临床研究,旨在评估本品与苯溴马隆的疗效差异。201名患者分别接受本品或苯溴马隆治疗,主要终点为试验结束时尿酸浓度相比基线值的变化,次要终点为尿酸达标率(低于6mg/dL)。结果显示本品2mg治疗组尿酸下降率为45.92%,而苯溴马隆组为43.87%,在治疗结束时,本品治疗组尿酸达标率为86.27%,而苯溴马隆组为83.27%,达到了临床意义上的非劣性标准[94]。试验二(NCT03372200)的设计与试验一类似,不过对照药物换成了非布司他。201名患者分别接受本品或非布司他治疗,本品2mg治疗组尿酸下降率为41.82%,而非布司他治疗组为44.00%,在治疗结束时,本品治疗组尿酸达标率为84.8%,而非布司他组为88.0%,也达到了临床意义上的非劣标准[95]。试验三(NCT03006445)的研究目的是探索长期用药的安全有效性,330名患者长达58周接受本品治疗,在34周时,接受2mg/天治疗患者的尿酸下降率为46.73%,接受4mg/天治疗患者的下降率为54.92%,尿酸达标率分别为89.11%和97.50%,而58周时,接受两种不同剂量治疗患者的尿酸下降率分别为47.17%和57.35%,尿酸达标率分别为91.30%和100%,总不良反应发生率为21.8%,主要为痛风性关节炎12.7%,关节炎2.1%和肢体不适2.1%[96]。随着生活水平的提高,痛风、高尿酸血症逐渐与高血压、糖尿病并列,是困扰人类健康的主要因素之一,但现有的产品几乎存在疗效不理想或安全性风险高的问题,治疗需求远远未得到满足,临床研究结果显示本品疗效较好,而且不良反应适中,是一个非常值得关注的产品。
Preview
来源: 药事纵横
3月25日,PMDA首次批准了日本新药株式会社的反义寡核苷酸viltolarsen,8月12日,该产品也获得了FDA的加速批准,用于DMD(杜氏肌营养不良症)基因53号外显子跳跃突变的杜氏肌营养不良症治疗。FDA批准本品上市是基于一项多中心、两阶段的剂量相关性研究(NCT02740972)的结果。16名受试者分别随机接受本品或安慰剂治疗4周,然后患者均接受两个不同剂量的viltolarsen进行开放标签治疗20周,以第25周肌营养不良蛋白水平相对于基线的变化情况来评价治疗效果。结果显示,接受本品80mg/kg每周一次治疗的8名患者,通过Western blot方法测定的平均肌营养不良蛋白水平从基线时正常值的0.9%升高至正常值的5.9%,而使用质谱法测定的平均肌营养不良蛋白水平从基线时正常值的0.6%升高至正常值的4.2%,达到预先设定的试验终点。DMD是一种X连锁的遗传病,整体患病率为2.8/10万,DMD患者的肌营养不良蛋白水平通常不足正常值的3%,大部分患者因呼吸衰竭和心脏功能障碍而死亡[97,98]。虽然此前FDA已经批准了eteplirsen、golodirsen和地夫可特等药物,但临床需求依然未得到良好的满足,EVP预测本品在2026年的销售额可达5.85亿美元。
3月25日,PMDA也批准了小野制药的tirabrutinib,用于复发或难治性的原发性中枢神经系统淋巴瘤、原发性巨球蛋白血症和淋巴浆细胞淋巴瘤治疗。Tirabrutinib 是一种第二代布鲁顿氏酪氨酸激酶(BTK)抑制剂[99],PMDA批准该产品是基于两项临床试验的结果,其中试验一(ONO-4059-02)是一项1/2期临床试验,17名复发或难治性的原发性中枢神经系统淋巴瘤患者接受治疗后,总缓解率为52.9%,其中完全缓解1例,部分缓解8例[100]。试验二(JapicCTI-173646)是一项针对原发性巨球蛋白血症和淋巴浆细胞淋巴瘤设计的临床试验,27名患者接受本品480mg每日一次治疗,18名初治患者亚组的总缓解率为88.9%(16名患者部分缓解),9名复发或难治的患者亚组的总缓解率为88.9%(8名患者部分缓解),总不良反应发生率为92.6%,主要为皮疹和中性粒细胞减少症[101]。除了获批的适应症外,本品在开发的适应症还包括弥漫性大B细胞淋巴瘤、套细胞淋巴瘤、慢性淋巴细胞淋巴瘤等,美国的临床试验正在有序推进中。
Preview
来源: 药事纵横
3月25日,PMDA还批准了Stella Pharma的borofalan B-10,用于不可切除、放化疗后的局部晚期或局部复发性头颈癌治疗。这是一种有别于放疗的新型疗法,简称硼中子捕获疗法(BNCT),作用机理是含10B的药品注射到患者体内,经载体选择性地携带入肿瘤细胞,然后通过中子照射激发10B核变,释放出带能量的α粒子和7Li原子杀死癌细胞。PMDA批准该产品上市是基于一项开放标签的2期临床试验(JapicCTI-194640)的结果,21例复发性鳞状细胞癌(R-SCC)或复发/局部晚期非鳞状细胞癌(R/LA-nSCC)患者接受本品500mg/kg的剂量输注后,相关部位接受C-BENS(回旋加速器超热中子源)照射。结果显示,71%的患者出现缓解,其中5例完全缓解,10例部分缓解,8名R-SCC患者的2年期生存率为58%,中位局部无进展生存期为11.5个月,而13名R/LA-nSCC的2年期生存率为100%,中位局部无进展生存期尚未达到,不良反应主要是脱发(95%),高淀粉酶血症(86%)和恶心(81%)[102]。这是一项非常值得关注的技术,临床试验不但展示了较高的有效性,而且相比普通化疗,其对正常细胞的损坏效果更小,可使用于放疗后复发的患者,还可以使用PET-CT监视治疗后的效果[103,104]。
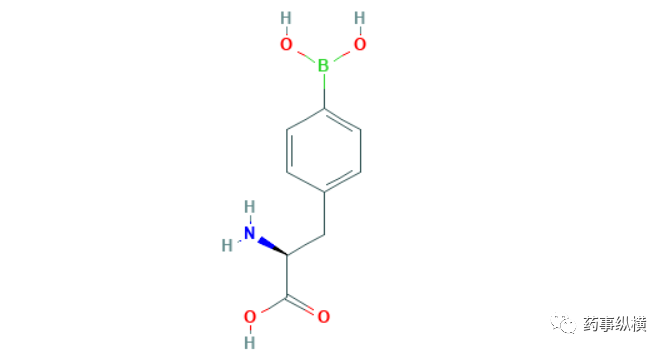
Preview
来源: 药事纵横
同在3月25日,PMDA批准的第四个药物为tepotinib,这是默克雪兰诺公司的原癌基因蛋白质c-Met抑制剂,用于MET基因外显子14跳跃突变的非小细胞肺癌MET基因外显子14跳跃突变的非小细胞肺癌治疗。PMDA批准本品上市是基于一项国际多中心、开放标签的2期临床研究结果。152名携带MET基因外显子跳跃突变、无法手术切除或复发的晚期非小细胞肺癌患者接受了本品治疗,其中99名患者完成了至少9个月的随访。结果显示总缓解率为46%,中位DOR为11.1个月,主要不良反应为外周水肿、恶心和腹泻[105],数据显示,约3%-4%的NSCLC患者携带MET基因外显子14跳跃突变[106],单凭这个适应症,tepotinib很难掀起肺癌市场的波澜,不过有文献报道该产品可以克服NSCLC癌细胞因c-Met激活而对EGFR(表皮细胞生长因子受体)抑制剂产生的耐药性[107],而且一项与吉非替尼联用的1期临床研究已初步探索到联合用药在c-Met高表达患者中的获益性[108]。
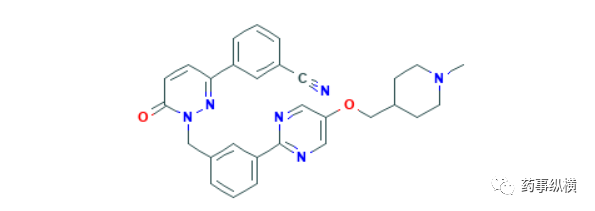
Preview
来源: 药事纵横
7. Veklury(瑞德西韦)
5月7日,PMDA首次批准了吉里德瑞德西韦,10月22日,FDA也批准了该产品,用于12岁及以上年龄段需要住院的2019年新冠病毒(COVID-19)感染者治疗。FDA批准瑞德西韦上市是基于三项临床研究的结果,试验一(NCT04280705)是由NIAID开展的临床试验,1062名轻中重度患者在标准护理的条件下接受瑞德西韦或安慰剂治疗,试验终点为29天内患者恢复(无需通氧及持续的医疗服务或患者出院)时间。结果显示,瑞德西韦治疗的541名患者,中位恢复时间为10天,而安慰剂组的521名患者,中位恢复时间为15天。两组轻中度患者中位恢复时间均为5天,而重度患者分别为11天和18天[109]。试验二(NCT04292899)是一项随机、开放标签、多中心的临床试验,该试验比较了重症患者分别接受瑞德西韦5天疗程(n=191)和10天疗程(n=200)的疗效差异。结果显示,在调整两组间基线差异之后,两组患者第14天的临床状态改善程度相似,死亡率和恢复率无显著差别[110]。试验三(NCT04292730)与试验二设计类似,该试验比较了患者分别接受瑞德西韦5天疗程(n=197)、10天疗程(n=193)和标准护理(n=200)后第11天的临床状态差异。结果显示,与仅接受标准护理的患者相比,瑞德西韦5天疗程组临床状态显著性改善,10天疗程组的改善程度未达到统计学显著性差异[111]。尽管瑞德西韦已经日本和欧盟批准,但该产品的疗效依然饱受争议[112],WHO通过大数据分析认为该产品对住院患者帮助甚微或没有帮助[113]。不过质疑归质疑,吉利德三季度财报显示,该产品在2020年前三季度的销售额就已达8.73亿美元,全年销售额极可能超过10亿美元。
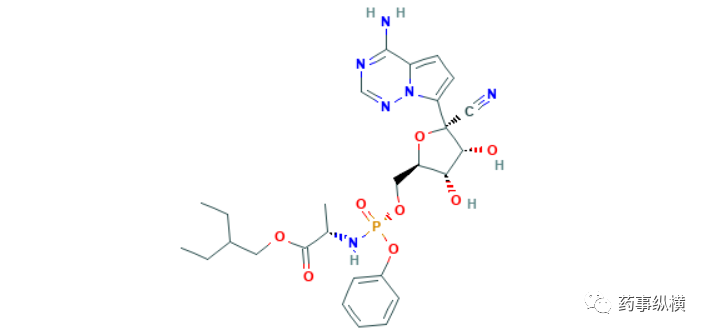
Preview
来源: 药事纵横
6月29日,PMDA首次批准了中外制药的IL-6R (白介素-6受体)单抗satralizumab,8月14日,该产品也获得了FDA批准,用于AQP4抗体阳性的视神经脊髓炎谱系疾病治疗。Satralizumab的获批是基于两项为期96周的临床研究结果。试验一(NCT02073279)入组了95名患者,其中64名患者AQP4为阳性,41名患者接受了本品治疗,另外23名接受了安慰剂治疗,试验二(NCT02028884)入组了76名患者,其中52名患者AQP4为阳性,26名患者接受了本品治疗,另外26名接受了安慰剂治疗。治疗结果显示,试验一中两组患者的NMOSD复发率分别为22.0%和56.5%,相比安慰剂,satralizumab治疗组复发风险降低了74%[114],而试验二的结果与试验一类似,两组患者的NMOSD复发率分别为11.5%和42.3%,相比安慰剂,satralizumab治疗组复发风险降低了78%,均达到统计学显著性差异。两项临床试验均无证据证明AQP4阴性的患者在治疗过程中有获益[115]。由于satralizumab是2020年第二个获批NMOSD的产品,市场预期相对第一款将有所下调,EVP预测该产品在2026年的销售额将达4.19亿美元。
6月29日,PMDA批准了三菱田边制药的vadadustat,用于CKD贫血治疗。Vadadustat是第二个获批上市的HIF-PH抑制剂,PMDA批准该产品是基于在日本开展的四项临床试验的结果。试验一是一项针对保守治疗期CKD患者开展的非劣性试验,受试分别使用本品或α达依泊汀(EPO)治疗52周。第20-24周的治疗结果显示,vadadustat组平均血红浓度为11.66g/dL(n=151),其中使用红细胞生成刺激剂(ESA)的患者亚组为11.43 g/dL(n=80),未使用ESA的患者亚组为11.88 g/dL(n=71),而EPO治疗组为11.93 g/dL(n=153),其中使用ESA的患者亚组为11.77 g/dL(n=82),未使用ESA的患者亚组为12.04 g/dL(n=71),本品与EPO达非劣性标准。第48-52周的结果显示,vadadustat治疗组血红蛋白浓度达标率为65.56%,其中使用ESA的患者亚组为60.0%,未使用ESA的患者亚组为71.8%,而EPO治疗组为78.43%,其中使用ESA的患者亚组为79.3%,未使用ESA的患者亚组为77.5%。试验二是针对正在使用ESA的血透患者设计的试验,其试验设计和试验结果与试验一类似。试验四是针对腹透患者开展的一项开放性标签的试验,41名平均血红蛋白浓度为10.89 g/dL的腹透患者,接受本品治疗20-24周,平均血红蛋白浓度上升至11.35 g/dL,在48-52周间测定血红蛋白浓度显示,达标率为64.3%[116]。在全球范围内,CKD的患病率为8–16%,而贫血是CKD最常见的并发症之一,五期CKD贫血患者比例高达90%[117]。HIF-PH抑制剂是一类疗效可以媲美促红素的口服疗法,市场潜力巨大,但遗憾的是大型临床试验数据暗示该产品的心脏安全性不及α达依泊汀,EVP下调了销售额预测,预测该产品在2026年的销售额为5.28亿美元。
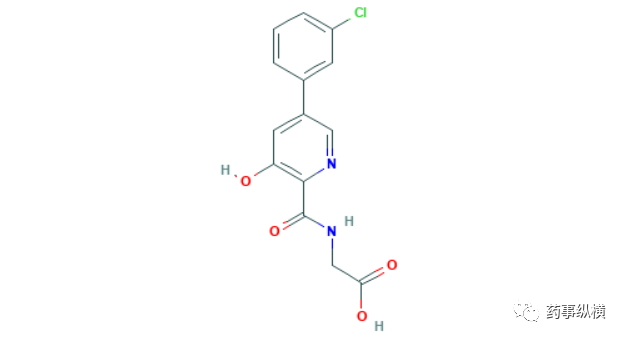
Preview
来源: 药事纵横
在批准vadadustat的同一天,还批准了它的竞品daprodustat,两者作用机制和获批适应症完全相同,daprodustat由GSK开发,PMDA批准该产品是基于在日本开展的三项临床研究的结果。试验一(NCT02791763)是一项非劣性试验,299名保守治疗期的CKD患者分别接受daprodustat和聚乙二醇β依泊汀治疗,第40-52周的治疗结果显示,daprodustat治疗组平均血红蛋白浓度为11.97g/dL,其中使用ESA的患者为12.00g/dL,而未使用ESA患者为11.90 g/dL,而β依泊汀治疗组为11.86 g/dL,其中使用ESA的患者为12.01g/dL,而未使用ESA患者为11.66g/dL,统计结果达非劣性[118]。56名腹透患者(其中3名未使用ESA)在40-52周的平均血红蛋白为12.09g/dL,所有患者的血红蛋白浓度均达到预期范围(11-13 g/dL)。试验二(NCT02969655)是一项针对使用ESA的血透患者设计的临床试验,试验设计与试验一类似,第40-52周的治疗结果显示,daprodustat治疗组平均血红蛋白浓度为10.89g/dL(n=133),而α达依泊汀治疗组为10.83g/ dL(n=134),统计结果达到非劣性标准[119]。试验三(NCT02829320)是一项针对不使用ESA的血透患者开展的临床试验,28名受试者在入组时平均血红蛋白浓度为9.10g/dL,治疗24周后,平均血红蛋白浓度上升至11.12g/dL,所有患者的血红蛋白浓度均达到预期范围(10-12 g/dL)[120]。由于大规模临床试验数据显示,vadadustat的心脏安全性不及α达依泊汀,这对GSK而言,少了一个主要竞争对手,EVP预测daprodustat在2026年的销售额可达8.90亿美元。
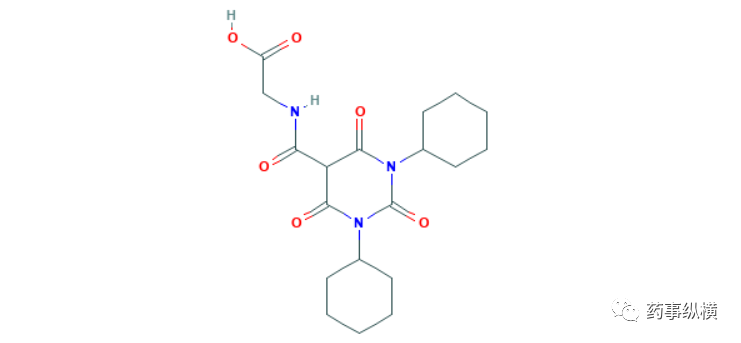
Preview
来源: 药事纵横
11. Enaroy(enarodustat)
9月25日,PMDA批准了日本烟草公司的enarodustat,这是第四个获批用于CKD贫血治疗的HIF-PH抑制剂。Enarodustat的获批是基于6项临床实验的结果,试验一是一项针对保守治疗的CKD患者开展的临床试验,216名患者分别接受enarodustat或α达依泊汀治疗,第20-24周的治疗结果显示,enarodustat组平均血红浓度为10.96g/dL(n=96),而α达依泊汀治疗组为10.87g/dL(n=96),达非劣性标准。试验二也是一项针对保守治疗的CKD患者开展的临床试验,132名患者接受本品治疗52周后,平均血红浓度为10.56g/dL,治疗结束时,所有患者的血红蛋白浓度维持在目标范围内(10-12 g/dL)。试验三是一项针对腹透患者开展的临床试验,42名患者接受本品治疗52周后,平均血红浓度为10.78g/dL,治疗结束时,所有患者的血红蛋白浓度也维持在目标范围内(10-12 g/dL)。试验四为一项针对正在试验ESA的血透患者开展的临床试验,173名患者分别接受本品或α达依泊汀治疗,第20-24周的治疗结果显示,enarodustat组平均血红浓度为10.73g/dL(n=78),而α达依泊汀治疗组为10.85g/dL(n=80)达非劣性标准。试验五是一项开放标签的临床试验,34名未使用ESA的血透患者,使用本品治疗后血红蛋白浓度上升速度为每周0.302g/dL,在治疗第8周时,血红蛋白浓度达到预定的范围(10-12 g/dL)。试验六是一项开放标签的临床试验,探索了使用ESA的血透患者的长期用药安全性。136名患者使用本品治疗52周,基线时血红蛋白浓度为10.61 g/dL,而治疗结束时为10.72g/dL,并维持在预期范围(10-12 g/dL)内,不良反应发生率为8.8%,主要不良反应为高血压和皮疹[121]。
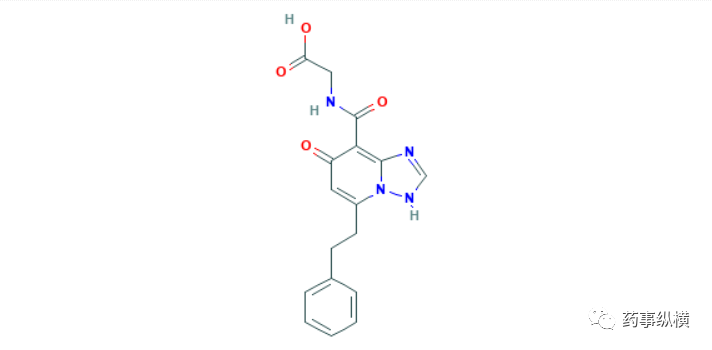
Preview
来源: 药事纵横
9月25日,PMDA还批准了科研制药株式会的sofpironium bromide凝胶,用于原发性腋窝多汗症治疗。Sofpironium bromide是一种胆碱受体阻断剂,PMDA批准该产品上市是基于两项临床试验的结果,其中试验一是一项安慰剂对照的双盲试验,而试验二是一项开放标签的试验。在试验一中,281名多汗症疾病严重程度量表(HDSS)评分为3级以上、且每个腋窝出汗量不低于50mg的原发性腋窝多汗症患者,分别接受本品或安慰剂持续治疗6周,结果显示,本品治疗组有53.9%的患者达到治疗终点(HDSS评分为1级或2级、两腋窝的出汗量下降不低于50%),而安慰剂组仅为36.4%。试验二入组的患者在入组时,均已经完成对照性试验研究,入组后全部接受本品持续治疗52周,结果显示57.8%的患者达到治疗终点(与试验一相同),主要不良反应是用药部位发炎、瘙痒、潮红、皮疹、瞳孔扩大,视力模糊等,相比试验一,试验二表现出的不良反应更多,发生率更高[122,123]。多汗症是一种非常常见的疾病,流行病学数据显示,美国人多汗症患病率为4.8%[124],德国患病率为16.3%[124],治疗需求巨大,在此之前,FDA曾批准格隆溴铵(Qbrexza)用药该病治疗,礼来年报数据显示,该产品在上市后的第一个完整财年的销售额就达2100万美元,如果本品能够在全球范围内上市,也极有可能成为一款年销售额超过1亿美元的产品。
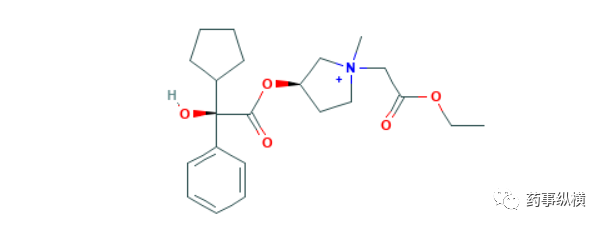
Preview
来源: 药事纵横
同在9月25日,PMDA还加速批准了Rakuten Medical的ADC药物,用于不可切除的局部晚期或局部复发性头颈癌治疗。PMDA批准该产品是基于一项1期临床试验和一项1/2期临床试验的结果。在试验一中,3名无法手术切除的颈部鳞状上皮细胞癌患者,在接受640 mg/m2剂量的Akalux输注后的20-28小时,再接受690nm的激光束照射治疗,结果显示,2名患者出现部分缓解。试验二与试验一的设计类似,30名患者接受治疗后,观察到的总缓解率为43.3%,其中4名患者完全缓解,13名患者部分缓解。整个试验中,25名患者出现了不良反应,主要不良反应为面部水肿,疲劳,红斑、吞咽障碍、舌头浮肿和咽喉肿痛[126]。Cetuximab是一种EGFR单抗,而sarotalocan是一种无毒的亲水性的硅-酞菁衍生物IRDye700DX,Akalux的获批之所以引人关注,是因为它是首个获批上市的光免疫疗法。Akalux的作用机理与传统的光动力疗法不同,其光敏剂在收到近红外光照射时,会失去亲水基团而导致溶解性改变,进而导致偶联物的聚集,这种聚集又可诱导细胞膜内压力改变,使得跨膜水流量增加,最终导致癌细胞破裂死亡。Akalux诱导的免疫原性细胞死亡过程中,死亡细胞附近的未成熟树突状细胞也会被诱导成熟,从而引发宿主抗癌免疫反应。这种疗法,不但可以增加药物对癌细胞的杀灭能力,还可以降低对正常细胞的影响,在实体瘤治疗方面,具有非常巨大的发展前景[127-129]。
药事纵横投稿须知:稿费已上调,欢迎投稿
图片广告:

Preview
来源: 药事纵横

Preview
来源: 药事纵横
药事纵横是一个开放,由自愿者组成的团体,由以下成员组成:Voyager88,雷道安,Herman,梅希,文竹,duke,子炎,ZMJ和曾文亮。欢迎有志之士加入我们团队。投稿、合作、加专业群请加微信437180999,药事纵横二千人QQ群22711855
来和芽仔聊天吧



立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。