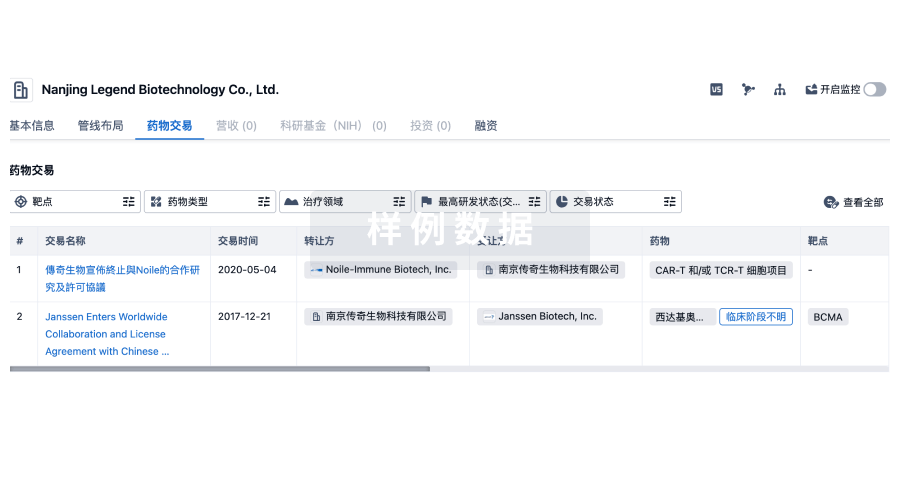
点击上方的 行舟Drug ▲ 添加关注摘要通过收集美国FDA网站公布的批准前检查相关法律法规,研究美国FDA药品批准前检查(preapproval inspection, PAI)的发展历程,重点对美国FDA药品批准前检查合规项目手册及其修订过程进行分析讨论。美国FDA基于风险和科学逐步完善批准前检查合规项目手册,以确保上市产品的安全有效和质量可控。建议在进行药品注册现场核查时,可从全生命周期的考量出发,确定核查要点,同时强化风险管理意识,加强交流和协作,提升药品注册核查的质量和效率。关键词:批准前检查;药品注册核查;沟通协作;风险管理;全生命周期管理为保证人民用药安全,确保上市产品的安全有效和质量可控,美国FDA从20世纪90年代开始进行注册批准前检查(preapproval inspection, PAI),至今已有30多年的历史,PAI由药品审评和研究中心(Center for Drug Evaluation and Research, CDER)发起,监管事务办公室(Office of Regulatory Affairs, ORA)执行。为了指导检查员开展药品生产设施的PAI,美国FDA制定了PAI合规项目手册(Compliance Program Guidance Manual 7346.832 Preapproval Inspections, CPGM7346.832)。自首次发布以来,随着行业整体水平的不断提升和科学认知的持续深入,美国FDA正不断优化内部机构,引入先进理念,并基于风险对CPGM 7346.832进行了多次重大修订,如根据2002年提出的基于风险的倡议,修订检查范围从全面检查过渡到基于风险的检查;根据数据完整性方面发现的问题,修订检查目标增加数据可靠性,从关注药品生产质量管理(current good manufacture practices, CGMP)合规性到更加关注产品本身;为提高审评和检查的效率,纳入2017年提出的综合质量评估(integrated quality assessment, IQA)团队;再到为应对复杂环境的影响,引入2021年提出的新的设施评估工具-远程交互评估(remote interactive evaluation, RIE),以及为了提高监管的科学性,增加合规目标“药品研发质量承诺”和全生命周期的考量等。采取多种措施以应对复杂多变的环境带来的挑战,确保按时限高效地完成PAI。本文通过对美国FDA PAI的介绍及对CPGM 7346.832的分析和讨论,以期为我国药品注册核查工作提供参考和借鉴。一美国FDA API 发展历程1.1 PAI的由来1984年,美国通过的《Hatch-Waxman法案》,又名《药品价格竞争与专利期补偿法》,在给予专利药专利期延长和法规独占期补偿的同时,也为仿制药提供了简化申请的途径,仿制药产业方兴未艾。在利益的驱使下,一些制药企业为抢首仿,不惜造假和/或编造申报资料数据。许多仿制药生产商未开展或采用参比制剂代替自制制剂进行体内生物等效性试验,行为极其恶劣。与此同时,部分制药企业为了独占市场,拖延竞争对手产品上市,贿赂美国FDA官员使其延迟或加速产品审批。为应对上述问题,美国FDA在中-大西洋地区首先实施了药品注册PAI,并于1990年10月发布了第1版CPGM 7346.832 PAI,自此结束了基于诚信的药品审批制度,取而代之的是除GMP合规性外,在药品注册申请批准前,美国FDA还会对递交的所有数据的充分性和准确性进行审查。1.2 PAI发展历程中的重大变革自CPGM 7346.832实施以来,药品注册PAI逐步发展为基于风险、从产品本身出发的全生命周期的管理。随着发展和理念的转变,CPGM 7346.832 PAI合规项目手册也在不断修订完善。以下主要从检索到的几次重大的修订来阐述其发展历程。与20世纪90年代中期相比,美国FDA收到的新分子实体和生物制品许可申请的数量急剧下降,而仿制药申请数量却在稳步上升,为了鼓励企业改变投资方向和强化创新,2002年美国FDA发起了一项为期2年的倡议,即“21世纪GMP,一种基于风险的方法”,鼓励使用基于风险和科学的监管方法提升产品质量,并促使监管现代化。2003和2004年修订的CPGM 7346.832很好地体现了这一倡议,并引入了基于风险的方法。修订后的合规项目手册给予了地区检查办公室更大的灵活性,使其可以根据企业的合规历史和申报情况等决定是否需要PAI。该版本的合规项目手册还删除了治疗窗窄的药品、最常用200种处方药的仿制药检查类别等,通过风险评估减少了PAI的数量。该版本的合规项目手册的检查目标主要围绕生产工艺、重新加工/返工、分析实验室、物料、厂房设施设备、包装和标签等几个方面,更关注CGMP合规性,不批准的情形也主要是严重偏离CGMP的问题或其他影响PAI的情形。2005年,美国FDA收到Able质量控制实验室人员的投诉举报,称Able Laboratories存在数据可靠性问题。该企业此前已接受过7次PAI,且均未发现任何伪造的证据。美国FDA通过对Able Laboratories电子数据的检查,发现其存在严重的数据可靠性问题。2010年5月实施的CPGM 7346.832修改了检查目标,除商业化生产条件与申报的符合性外,还增加了数据可靠性。同时该版本的合规项目手册继续完善基于风险的策略,优化了评估项目,采用决策树进行风险评估以判定是否需要开展PAI。该版本的合规项目手册还细化了不批准的情形,除CGMP合规性外,同时关注品种本身的研究情况,如关键临床或申报批次的设备或工艺参数与注册批所用工艺的差异,申请中承诺事项的执行情况、方法学验证或确认过程的问题会引发产品或API稳定性的问题,或与稳定性研究相关的重大失败情况等。与2005年修订版本相比,该版本的合规项目手册有了重大的变化,也是后续版本修订的基础。随着制药行业的不断发展,审评审批中存在的问题也日益突出,CDER和ORA的交流不畅及反馈程序复杂,使得检查或重新检查后合规状态的更新速度缓慢,进而影响了产品批准与上市的时间。2017年9月,美国FDA发布了《人用药设施评估和检查计划综合运营方针(ConOps)》,组建由审评和现场检查人员共同组成的“综合质量评估(integrated quality assessment, IQA)”团队,以更加全面、细致地考虑所有可能的风险,通过加强协调沟通机制以解决悬而未决的质量问题。此外,ConOps还概述了CDER和ORA在人用药品设施评估和检查中的角色和职责。2019年修订的CPGM 7346.832突出了美国FDA项目整合和机构改革方面的变化,纳入了IQA团队,明确了工艺和设施办公室(Office of Pharmaceutical Quality, OPF)及药品质量办公室(Office of Pharmaceutical Quality, OPQ)等新机构在PAI中的职能以及ORA的新工作职能等。该版本合规项目手册引入了数字化的理念,通过信息化平台panorama进行风险评估,削减了人为因素可能带来的影响,并增加了潜在官方行动指示(pOAI)的报告职责,明确指出如果检查范围包括CPGM7356.002,即涉及已上市产品,并且发现可能导致官方行动的指示,应尽快在Panorama平台中录入pOAI警示,以便及时采取措施,将风险有效传递至已上市品种,从而及时有效地降低可能给患者带来的风险。受全球新型冠状病毒肺炎疫情流行等因素影响,2020年起美国FDA监管效率降低。对制药企业,尤其对生产API和制剂的工厂产生了重大的影响,甚至影响了药品的可及性。美国政府问责办公室呼吁美国FDA考虑现场检查的替代方法以解决积压问题。2021年美国FDA发布了《设施远程评估指南》,2022年9月美国FDA再次修订CPGM 7346.832。本次新修订的版本提出了新的设施评估工具——RIE,合规目标增加了“药品研发质量承诺”,引入了ICH Q12药品生命周期管理的理念和考虑,凸显了对企业全生命周期中风险识别、评估和控制能力的高度关注。二分析与讨论2.1 检查目标和范围随着行业的进步,CPGM 7346.832的检查目标也在不断适配修订,重心由CGMP合规性过渡到产品本身,并基于风险确定PAI的检查范围,开展针对性的检查(见表1)。CPGM 7346.832在每个检查目标项下列举了包括检查思路、重点、可能存在的问题及检查依据等方面的建议。如目标3数据可靠性,建议从关键数据出发,关注异常事件及企业采取的措施,分析引起数据可靠性的原因,进而发现企业在风险识别、分析和控制上的不足,窥一斑而知全豹。▲表1-美国FDA PAI检查目标变更历史为适应医药行业不断发展的需要,国内药品注册核查的目标也在不断迭代修订。根据我国新版《药品注册管理办法》,注册核查是指核实申报资料的真实性、一致性以及药品上市商业化生产条件,具体细化为研制现场的9个核查要点和生产现场的6个核查要点。为使药品注册核查工作高效化、风险可控、更好地为审评提供技术支撑,国家药品监督管理局食品药品审核查验中心(以下简称核查中心)也在不断探索尝试新的模式,基于药品注册核查的目标,通过风险分析确定被核查单位、核查方式及核查要点。在现场核查时,可根据被检查单位的合规历史、品种因素、场地因素等确定核查要点。通过发现的问题,延伸扩展至企业在风险识别、分析和控制等方面可能存在的不足,有的放矢,增强核查的针对性,提升药品注册核查的效率。2.2 检查方式受全球新型冠状病毒肺炎疫情的影响,2022年10月实施的CPGM 7346.832增加了RIE的检查方式,以期应对形式复杂的环境变化。某些情况下,美国FDA可能会选择RIE或在PAI之前进行RIE,RIE的结果可用于支持上市申请和补充申请,为按时限完成PAI提供了多一层保障。在新型冠状病毒肺炎疫情防控期间,核查中心也创新性地利用中国制度的优势,采用了联合各省级药品监督管理局的方式共同开展药品注册核查。同时采用远程非现场的方式对部分品种进行了境外非现场检查,通过检查对JW Life Science Corporation的脂肪乳氨基酸(17)葡萄糖(11%)注射液、UCB Pharma S.A.的左乙拉西坦注射用浓溶液和GlaxoSmithKline(Ireland)Limited的度他雄胺软胶囊采取了暂停进口、销售和使用的处理措施。特殊时期,对督促国外药品上市许可持有人和生产企业遵守我国的法律法规和技术标准起到了积极的促进作用。为提高药品注册核查的抗风险能力和核查效率,可结合境外非现场检查的经验,结合上市后监管,通过申请人提交的申报资料提前了解品种特性,同时结合被核查单位的合规历史等提前介入,识别和确定可能存在的风险及现场核查时需重点关注的内容,在药品注册任务繁重而核查资源不足的情况下提升药品注册核查的质量。2.3 结果判定美国FDA的合规项目手册中列明了应签发483表格的缺陷项和判定为不通过的情形,指导检查员对发现的问题的严重情况进行分析和认定。对于需CDER进一步评估的情形或问题,ORA会给出暂不批准的建议,待企业通过有效的纠正与预防措施实现对风险的控制后,可能会更新PAI的建议,以促使企业更加重视存在的问题和不足,积极改进。国内的核查要点及判定原则列举了需要关注的内容以指导药品注册核查,针对注册核查中存在严重偏离药品生产质量管理规范等相关法律法规的情形,或存在严重的数据可靠性等问题且导致对药品质量评价产生影响时,认定为“不通过”。在现场核查过程中,可参考《药品生产现场检查风险评定指导原则》中的缺陷定义和列举以及CPGM 7346.832中提出的问题类型及级别判定,根据国内的核查要点及判定原则给予最终的结论。2.4 风险传递风险的识别、评估、控制、审核和沟通是质量风险管理程序的关键内容。如何通过有限的监管识别的风险来确保风险的控制是基于风险开展核查的核心所在。根据CPGM 7346.832,检查组在进行PAI时,如发现企业在CGMP合规性和数据可靠性等方面存在重大问题,可扩大检查范围至CPGM 7356.002药品生产检查涵盖的内容。如果在扩大检查期间发现了上述问题,提示该场地生产的所有产品均有潜在风险,ORA会建议CDER启用注册诚信政策(AIP)或者建议制定有因检查计划以进一步明确发现的问题及涉及范围。如发现在投诉处理和上报规程方面存在重大问题,可考虑根据CPGM 7353.001对上市后不良反应报告系统进行检查。根据PAI发现的问题采取扩大检查范围等措施,以传递并确认企业可能存在的系统性风险。国内药品注册核查过程中,如果发现企业有违法、违规的情况,应及时移交当地药品监管部门处理,并在报告中予以专项说明。核查结束后,核查中心会通过“药品注册现场核查问题表”将检查组发现的问题及风险传递给省级药品监督管理局,使省级药品监督管理局除通过日常监管外,还可借助药品注册核查来识别辖区企业潜在的风险,监督企业及时落实整改措施。核查中心综合现场核查报告及省级药品监督管理局监督落实的整改情况等,将注册核查情况和核查结果反馈给国家药品监督管理局药品审评中心(简称药审中心),并提醒药审中心重点关注尚需进一步完善的问题,从而将药品注册核查的风险传递给日常监管的省级药品监督管理局和负责审评的药审中心,实现对已上市产品和其他在审品种的风险控制。药品注册核查发现的偏离生产质量管理规范的情形,如无合理解释而弃用记录和数据,或以其他方式选择性地使用记录和数据等问题,提示企业在整个质量管理体系方面可能存在不足,尤其是非品种因素的不通过情形,通常反映了已上市品种可能存在的严重风险,在现场核查过程中应予以重点关注,从而将单次核查识别的风险有效传递至被核查单位的其他品种,通过有限的监管辐射更大的范围,为基于风险的核查模型提供可靠有效的依据。2.5 沟通交流ORA和CDER在PAI的全流程上始终保持良好的沟通交流机制。根据ConOps,CDER在收到药品注册上市申请后会组建IQA团队,该团队包括审评员、CDER药品生产评估办公室(Office of Pharmaceutical Manufacturing Assessment, OPMA)生产评估员和ORA代表等。在开展PAI前,IQA团队以患者为中心,评估可能影响安全性和有效性的风险,并据此建议开展PAI的必要性。OPMA 生产评估员收集来自IQA团队关于现场处理这些问题的见解和建议,并以书面形式传递给ORA的批准前项目经理(preapproval program managers, PAMs)和检查员,以便其制定个性化的检查方案。针对检查中发现的问题,检查员通常可直接向OPMA生产评估员以及ORA的PAM提出。在检查结束后,IQA团队通过审核检查报告、483表格、企业整改回复、检查组建议等内容,确定影响注册申请审批的关键问题并提出建议。CDER综合评估后将最终的批准建议和考量反馈给ORA,以便于ORA更好地了解审评关注点,使检查更有针对性。CPGM 7346.832 附录中还列举了CDER和ORA在不同阶段的协作模式,指导团队成员开展有效的沟通以解决发现的问题。审评、核查和日常监管的密切沟通有助于核查前的风险识别、核查中的问题判定及核查后的反馈和落实。药审中心将识别的风险传递给核查中心,有利于核查中心基于风险确定核查场地、核查范围和核查要点,快速制定个性化核查方案,使检查组做到有的放矢,开展针对性的核查。核查过程中,良好的沟通可为检查组针对发现的重大问题或疑问提供更多的思路和解决途径,也便于监管部门及时采取措施。核查结束后,核查中心将需要提醒审评关注的内容反馈给药审中心,尤其是不通过的情形,以助力审评,使有限的资源得到最大化地利用。同时药审中心对核查发现问题的反馈可助力于核查中心加深核查发现问题对审评影响程度的把握和判定,适配核查的方向和内容,更好地发挥“眼睛”的作用。2.6 全生命周期的考量新修订的 CPGM 7346.832还引人了全生命周期管理的理念,以期减少因上市后变更而发起的PAI数量,使监管顺应行业的发展,更加科学化。如在目标1商业化生产条件中引入了既定条件(established conditions, EC);目标2与申报的符合性中,增加了风险管理系统实施的确认,确保在整个产品生命周期中能持续有效地识别、评估、处理发现的风险(例如:交叉污染;掺杂;有害杂质如亚硝胺、亚消化剂、亚硝酸盐、硝酸盐和叠氮化物)并将其传递给企业的管理层和美国FDA;引入了目标4药品研究质量承诺,美国FDA可借此收集信息用于数据分析或内部趋势分析,助力PAI风险因素的识别。美国FDA指出成熟先进的药品质量体系(pharmaceutical quality system, PQS)是持续合规及生产质量可控的保证。新修订的合规项目手册允许美国FDA评估企业PQS的某些方面,并深入了解企业用于持续系统改进的既定流程,注重风险的识别和控制的能力,关注研发阶段的设计和考量等,为减少上市后变更的监管提供有力的支撑。我国已于2017年加入了ICH,在药品注册核查时可借助ICH Q8药物研发、ICH Q9质量风险管理、ICH Q10药物质量体系(PQS)和ICH Q12药品生命周期管理的技术和监管考虑等指导原则提出的理念,关注研发阶段的设计和考量等,通过发现的问题进一步扩展、判定企业在风险识别、评估和控制方面的能力,为减少对已上市产品变更的监管累积数据。三思考和启示CPGM 7346.832自首次发布以来一直在不断修订和适配中,每次的修订均体现了监管部门对已暴露问题的态度、对可能发生问题的关注和重视以及监管科学性等方面的考量。国内的药品注册核查同样如此,齐齐哈尔第二制药有限公司假药事件、安徽华源公司欣弗事件、药物临床试验数据自查核查等,不仅促使了注册核查相关程序等的修订和调整,也凸显了注册核查的重要性。为了应对药品医疗器械审评审批中存在的问题,国务院办公厅印发了关于建立职业化专业化药品检查员队伍的意见,提出要着眼于药品监督检查工作体系的建立和检查员的培养。同时,为了加强国际合作,与国际接轨,2021年9月国家药品监督管理局正式致函药品检查合作计划(PIC/S),申请启动预加入程序,多措并举,助力国内药品检查能力的提升。药品注册核查本身也是质量风险管理的过程,为确保核查的力度和效力,开展药品注册核查时,可吸取过往的经验,学习国内外监管机构或组织发布的现场核查相关的合规手册和指南文件等,从全生命周期的角度出发,增加风险识别、评估和控制的能力,同时加强与各方的沟通交流,实现风险的有效传递,提升核查的力度、质量,确保按时限、科学、高效地完成药品注册核查任务,保障人民的用药安全。(后台回复关键词:2024425CDE文章 ▶ 获取PDF文件)参考文献[1] FDA.Complianee progmm 7346.832preapprval inspections[EB/0L].(2022-09-16)[2022-10-17].https://www.fda. gow/media/121512/download.[2] MARTIN D.HYNFS Ⅲ,药品注册批准前检查:美国药品监管法规核心理念概述(第2版)[M].北京:北京大学医学出版社,2011:1.9-11.[3] Able Laboratories,Ie. Cmnbemy, NJ, FDA 483 InspeetionalObservations, dated 05/02-07/01/2005[EB/0L1.(2005-07-06)[2022-10-17 ]. https ://www. fda. gov/media/70711/download.[4] FDA.Compliance progmm 7346.832 preappmval inspections[EB/0L1.(2010-04-12)[2022-05-121.https://www.fda. gow/mediw121512/download.[5] FDA. Integration of FDA facility evaluation and inspeetion pro.gmam for human dnugs : a coneept of opemations[EB/0L].(2017-06-06)[2022-10-171.https://www.fda. gov/dngs/phanma-ceutical-quality-resourees/integration-fda-facility-ewaluation-and-in.spection-program-human-dngs-concept-operations.[6] FDA. Remote interactive evaluations of dmg manufacturing andbioresearch monitoring facilities during the covid.19 publie healthemergeney guidance for industry[EB/0L].(2021-04-14)2022-10-171.https://www. fda. gow/regulatory-infonmation/search-fda-guidance-documents/remote-interactive-evaluations-dngmanufacturing-and-bioresearch.monitoring-facilities-during-covid.[7] FDA. Pharmaceutical CGMPS for the 2lst century-a risk-basedappmach final repon[EB/0L].(2004-09)[2022-10-17]https://www. fda. gov/media/77391/download.[8] FDA.Compliance progrm 7346.832 preappmval inspections[EB/0L].(2019-09-16)[2022-10-17].htps://www.fda.gov/dmugsguidance-compliance-regulatory-information/dnugcompliance-pngmms.[9] 国家市场监督管理总局.药品注册管理办法(局第27号)[EB/0L].(2020-01-22)[2022-10-17].https://gkml.samr.gov.cn/nsjg/fs/202003/t20200330 313670.htl.[10] 国家药品监督管理局食品药品审核查验中心.《药品注册核查工作程序(试行)》等5个文件的通告(2021年第30号)[EB/0L].(2021-12-20)[2022-10-17]. https://www.cfdi. org.en/resoure/news/14200. html.[11] 国家药品监督管理局.国家药监局关于暂停进口、销售和使用 JW LIFE SCIENCE CORPORATION 脂肪乳氨基酸( 17)葡萄糖(11%)注射液的公告(2022 年第 14 号)[EB/0L].(2022-01-29)[2022-10-17].hitps://www.nmpa. gov.en/xxgk/ggtg/qtggtg/20220129100413143. html.[12] 国家药品监督管理局.国家药监局关于暂停进口、销售和使用比利时 UCB Phama s.A.左乙拉西坦注射用浓溶液的公告(2022年第67号)[EB/0L].(2022-08-24)[2022-10-17].https://www.nmpa. gov.en/yaopin/ypggtg/20220824091021 196. html.[13] 国家药品监督管理局.国家药监局关于暂停进口、销售和使用 ClaxoSmithKline(Ireland)limited 度他雄胺软胶囊的公告(2022年第96号)[EB/0L].(2022-10-31)[2022-10-17]. https ://www.nmpa. gov.cn/xxgk/ggtg/qtggtg/20221031142605125. html.[14] 全国人民代表大会常务委员会,中华人民共和国药品管理法EB/0L].(2019-08-26)[2022-10-17 ]. httyp://www.gov.cn/xinwev2019-08/26/content 5424780.htm.[15] 原国家食品药品监督管理总局.《药品生产现场检查风险评定指导原则》(食药监药化监[2014]53号)[EB/0L].(2014-05-13)[2022-10-17].https://www. nmpa. gov.cn/xxgk/fg-wj/gzwygzwjyp/20140513120001350. html.[16] 原国家食品药品监督管理总局,国家食品药品监督管理总局关于开展药物临床试验数据自查核查工作的公告(2015 年第117号)[EB/0L].(2015-07-22)[2022 -10 - 17 ]. ht-tps://www.nmpa. gov.en/yaopin/ypggtg/ypqtgg/20150722173601172. html.[17] 国务院办公厅.国务院办公厅关于建立职业化专业化药品检查员队伍的意见(国办发[2019]36号)[EB/OL1.(2019-07-18)[2022-10-171.http://www.gov.cn/zhengeecontent/2019-07/18/content 5411172. him.[18] 国家药品监督管理局,国家药品监督管理局启动药品检查合作计划(PIC/S)预加人申请工作[EB/0L1.(2021-09-29)2022-10-171.hitps ://www.nmpa. gov.cwyaowen/ypjgyw/20210929162552105.html.[19] 叶笑,董江萍,化学药创新药注册核查主要问题分析及对策[J].中国新药杂志,2023.32(12):1206-1211.[20] 胡小娟,肝素类药品生产现场核查常见问题分析与探讨[J1.中国医药工业杂志,2023.54(4):617-621.[21]颜若曦.从注册核查视角对药品研制质量管理要点的探讨[J].中国新药杂志,2024.33(1):28 -35.文章信息源于公众号注册圈,登载该文章目的为更广泛的传递行业信息,不代表赞同其观点或对其真实性负责。文章版权归原作者及原出处所有,文章内容仅供参考。本网拥有对此声明的最终解释权,若无意侵犯版权,请联系小编删除。学如逆水行舟,不进则退;心似平原走马,易放难收。行舟Drug每日更新 欢迎订阅+医药大数据|行业动态|政策解读