预约演示
更新于:2025-08-04
Panitumumab
帕尼单抗
更新于:2025-08-04
概要
基本信息
药物类型 单克隆抗体 |
别名 Panitumab、Panitumumab (Genetical Recombination)、Panitumumab (genetical recombination) (JAN) + [12] |
靶点 |
作用方式 拮抗剂 |
作用机制 EGFR拮抗剂(表皮生长因子受体erbB1拮抗剂) |
在研适应症 |
原研机构 |
最高研发阶段批准上市 |
首次获批日期 美国 (2006-09-27), |
最高研发阶段(中国)申请上市 |
特殊审评优先审评 (美国)、加速批准 (美国) |




登录后查看时间轴
结构/序列
Sequence Code 103929L

来源: *****
Sequence Code 194636H
来源: *****
关联
326
项与 帕尼单抗 相关的临床试验NCT06998940
Randomized Phase III Study of Second-Line Chemotherapy With or Without Panitumumab for KRAS Wild Type, Locally Advanced or Metastatic Pancreatic Adenocarcinoma
NCT07094113
A Phase 1/1b Study Evaluating the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Efficacy of AMG 410 Alone and in Combination With Other Agents in Participants With KRAS Altered Advanced or Metastatic Solid Tumors
NCT07094204
A Phase 1 Study of ASP5834 in Participants With Locally Advanced (Unresectable) or Metastatic Solid Tumor Malignancies With KRAS Mutations or KRAS Amplifications

100 项与 帕尼单抗 相关的临床结果
登录后查看更多信息
100 项与 帕尼单抗 相关的转化医学
登录后查看更多信息
100 项与 帕尼单抗 相关的专利(医药)
登录后查看更多信息
2,471
项与 帕尼单抗 相关的文献(医药)2025-12-01Journal of Gastrointestinal Cancer
Anti-EGFR Rechallenge in Metastatic Colorectal Cancer and the Role of ctDNA: A Systematic Review and Meta-analysis
Review
作者: da Silva, Luís Felipe Leite ; Peixoto, Renata D 'Alpino ; Saldanha, Erick Figueiredo ; da Conceição, Lucas Diniz ; Noronha, Mariana Macambira ; da Silva, Marcos Vinícius Martins Grangeiro
BACKGROUND:
Metastatic colorectal cancer (mCRC) remains a significant clinical challenge. While anti-EGFR inhibitors have improved survival rates, their long-term efficacy is limited by disease progression, which is often associated with the development of acquired resistance mutations. However, some patients may regain sensitivity to anti-EGFR agents after alternative therapies, suggesting a potential benefit for rechallenge strategies. Our study aims to conduct a systematic review and meta-analysis to comprehensively evaluate the efficacy and safety of EGFR rechallenge in patients with mCRC.
METHODS:
A systematic search of the MEDLINE, EMBASE, and Cochrane databases was conducted between October 28 and December 24, 2023, to identify clinical trials investigating treatment regimens incorporating panitumumab or cetuximab as a rechallenge strategy. Pooled proportions or hazard ratios (HR) were calculated using a random effects model. Inter-study heterogeneity was assessed using the I2.
RESULTS:
Among the 2105 articles identified through the search, 13 met the predetermined inclusion criteria. Of these, 12 were phase II studies, encompassing 92.3% of the patient population. Cetuximab was administered to 302 patients (75.1%), whereas panitumumab was utilized in 100 patients (24.9%).A pooled analysis of eight studies demonstrated an objective response rate of 20.50% (95% CI 7.94 to 33.07) and a disease control rate of 67.35% (95% CI 58.60 to 76.09). The median progression-free survival was estimated at 3.5 months (95% CI 2.68-6.69), with a median OS of 9.8 months (95% CI 6.71-12.89). Patients exhibiting RAS wild-type status in circulating tumor DNA (ctDNA) analysis derived enhanced benefits from anti-EGFR rechallenge (HR: 0.41; 95% CI 0.28-0.60, I2 = 60%). Common grade 3 or higher treatment-related adverse events included neutropenia (22.8%) and rash (14.9%).
CONCLUSION:
This meta-analysis underscores the efficacy and safety of anti-EGFR rechallenge as a promising therapeutic approach for a subset of patients afflicted with mCRC. The observed correlation between wild-type RAS status, as determined through ctDNA analysis, and improved OS signals the prospect of precision oncology in guiding treatment decisions.
2025-12-01Journal of Gastrointestinal Cancer
Final Results of ERBIMOX: A Randomized Phase II Study of Modified FOLFOX7 With or Without Cetuximab as First-Line Treatment for KRAS Wild-type Metastatic Colorectal Cancer
Article
作者: Zirrgiebel, Ute ; Depenbusch, Reinhard ; Müller, Lothar ; Lerchenmüller, Christian ; Boller, Emil ; Niemeier, Beate ; Marschner, Norbert ; Sahm, Stephan ; Potthoff, Karin ; Tesch, Hans
BACKGROUND:
The combination of FOLFOX/FOLFIRI with an EGFR-antibody (cetuximab/panitumumab) is a first-line standard for RAS wild-type metastatic colorectal cancer (mCRC). The OPTIMOX stop-and-go regimen, which reduces oxaliplatin-induced neuropathy, and fluorouracil/folinic acid (FU/FA) were standard maintenance-therapies in the pre-antibody era. Whether an EGFR-antibody adds value to the OPTIMOX strategy in the RAS wild-type setting remains unknown.
METHODS:
In the open-label, randomized, multicenter phase II ERBIMOX trial, patients with KRAS wild-type mCRC received either first-line induction-therapy with 8 cycles of mFOLFOX7 followed by maintenance-therapy with FU/FA (OPTIMOX arm) or mFOLFOX7 + cetuximab followed by FU/FA + cetuximab (ERBIMOX arm). Primary objective was to demonstrate superiority of additional cetuximab to mFOLFOX7 during induction/maintenance-therapy. Primary endpoint was objective response rate (ORR). Secondary endpoints included progression-free survival (PFS), overall survival (OS) and safety. The trial is registered at EudraCT (No.2006-002744-28).
RESULTS:
From 2006-2011, 138 patients with KRAS wild-type mCRC from 23 German sites were randomly assigned to either OPTIMOX (N = 63) or ERBIMOX (N = 75). ORR numerically favored the ERBIMOX arm (64.0% vs. 54.0%, P = 0.3071). Median PFS (ERBIMOX vs. OPTIMOX) was 9.6 vs. 8.8 months (P = 0.7612), median OS 25.6 vs. 30.9 months (P = 0.5821). Most common grade 3/4 adverse events (AEs) were skin reactions (21.9% vs. 2.1%) and gastrointestinal disorders (13.5% vs. 9.5%). No cetuximab-related deaths occurred.
CONCLUSION:
In treatment-naïve KRAS wild-type mCRC, adding cetuximab to mFOLFOX7 resulted in numerically higher ORR than mFOLFOX alone, but no statistically significant differences in ORR, PFS or OS; probably because of the premature stop due to poor recruitment. The safety profile was as expected, with few discontinuations.
2025-12-01Journal of Gastrointestinal Cancer
Efficacy and Safety of Anti-EGFR Therapy Rechallenge in Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis
Review
作者: Burbano, Rommel Mario Rodríguez ; de Oliveira Rodrigues, Anna Luíza Soares ; de Moraes, Francisco Cezar Aquino ; Limachi-Choque, Jhonny ; Priantti, Jonathan N
BACKGROUND:
Colorectal cancer (CRC) represents the second leading cause of cancer-related mortality worldwide, with a significant portion of patients presenting with metastatic disease at diagnosis. Resistance to initial anti-EGFR therapy, a key treatment for RAS wild-type metastatic CRC, remains a major challenge. This study aimed to assess the efficacy and safety of rechallenge with anti-EGFR therapy in patients with metastatic CRC who have progressed after prior treatments.
METHODS:
A systematic search was conducted across PubMed, Web of Science, Cochrane, and Scopus. Studies were included if they were randomized controlled trials (RCTs) or observational studies involving patients with EGFR-mutated metastatic CRC who received anti-EGFR therapy as a rechallenge. Endpoints included objective response rate (ORR), disease control rate (DCR), and the incidence of adverse events. Statistical analyses were performed using the DerSimonian/Laird random effect model, with heterogeneity assessed via I2 statistics. R, version 4.2.3, was used for statistical analyses.
RESULTS:
Fourteen studies were included with 520 patients; 50.3% were male, and the median age was 63 years old. The median progression-free survival (mPFS) ranged between 2.4 and 4.9 months, while the median overall survival (mOS) ranged from 5 to 17.8 months. Our pooled analysis demonstrated an objective response rate (ORR) of 17.70% (95% CI, 8.58-26.82%) and a disease control rate (DCR) of 61.72% (95% CI, 53.32-70.11%), both with significant heterogeneity (I2, 84% and 80%, respectively; p < 0.01). In the subgroup analysis, cetuximab showed an ORR of 18.31% (95% CI, 4.67-31.94%), and panitumumab an ORR of 10.9% (95% CI, 0.00-26.82%), while the combination of both resulted in an ORR of 29.24% (95% CI, 0.00-65.84%). For DCR, cetuximab resulted in 62.1% (95% CI, 49.32-74.87%), panitumumab in 63.05% (95% CI, 52.13-73.97%), and the combination in 60.34% (95% CI, 31.92-88.77%), all with significant heterogeneity. Adverse events included anemia (15.39%), diarrhea (4.20%), hypomagnesemia (6.40%), neutropenia (22.57%), and skin rash (13.22%).
CONCLUSIONS:
Rechallenge with anti-EGFR therapy in metastatic CRC patients shows moderate efficacy with manageable safety profiles. These findings highlight the need for careful patient selection and monitoring to optimize outcomes. Further studies are warranted to refine strategies for maximizing the therapeutic benefits of anti-EGFR rechallenge.
100 项与 帕尼单抗 相关的药物交易
登录后查看更多信息
研发状态
批准上市
10 条最早获批的记录, 后查看更多信息
登录
| 适应症 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|
| KRAS G12C突变结直肠癌 | 美国 | 2025-01-16 | |
| KRAS 野生型结直肠癌 | 美国 | 2014-05-23 | |
| 结直肠癌 | 日本 | 2010-04-16 | |
| RAS 野生型结直肠癌 | 澳大利亚 | 2008-05-14 | |
| 转移性结直肠癌 | 美国 | 2006-09-27 |
未上市
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 大肠腺癌 | 临床3期 | 美国 | 2018-02-27 | |
| 遗传性非息肉病性结直肠癌1型 | 临床3期 | 美国 | 2018-02-27 | |
| NRAS 野生型结直肠癌 | 临床3期 | 日本 | 2015-05-29 | |
| 转移性食管鳞状细胞癌 | 临床3期 | 德国 | 2012-05-01 | |
| KRAS 突变结直肠癌 | 临床3期 | 美国 | 2010-02-02 | |
| KRAS 突变结直肠癌 | 临床3期 | 澳大利亚 | 2010-02-02 | |
| KRAS 突变结直肠癌 | 临床3期 | 比利时 | 2010-02-02 | |
| KRAS 突变结直肠癌 | 临床3期 | 加拿大 | 2010-02-02 | |
| KRAS 突变结直肠癌 | 临床3期 | 捷克 | 2010-02-02 | |
| KRAS 突变结直肠癌 | 临床3期 | 法国 | 2010-02-02 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
临床2期 | RAS/BRAF wild-type | circulating DNA (cirDNA) | - | Panitumumab + FOLFIRINOX | 範蓋範廠鏇窪餘鏇鬱鹽(構顧糧襯構鏇遞醖糧製) = 鬱鬱鏇醖願鑰鏇襯鹹製 網選網築選廠廠淵觸蓋 (構窪膚簾窪襯夢糧顧壓 ) 更多 | 不佳 | 2025-07-05 | |
Panitumumab + mFOLFOX6 | 範蓋範廠鏇窪餘鏇鬱鹽(構顧糧襯構鏇遞醖糧製) = 膚選鬱構糧廠鏇鏇願鏇 網選網築選廠廠淵觸蓋 (構窪膚簾窪襯夢糧顧壓 ) 更多 | ||||||
N/A | 43 | 淵獵範願廠顧鹹鹹範夢(艱鏇鹹餘範顧製糧衊積) = 7% 窪膚顧糧夢鹽鹹醖鑰繭 (齋觸艱構繭膚廠願衊窪 ) 更多 | 积极 | 2025-07-03 | |||
N/A | 20 | 衊顧憲鏇獵構獵觸網構(鹹窪範淵顧繭壓構積範) = 5% 衊壓簾觸壓窪齋廠繭顧 (壓製糧壓醖積築憲餘獵 ) 更多 | 积极 | 2025-07-03 | |||
临床3期 | KRAS G12C突变结直肠癌 KRAS G12C | 160 | 廠獵鹽遞築艱鹽鬱繭構(鹹顧蓋選廠廠鹹築積願) = 鏇範齋憲顧窪蓋鬱選觸 窪艱獵獵鹽鑰襯鑰憲夢 (觸願鹹膚窪餘網願製糧 ) 更多 | 积极 | 2025-05-07 | ||
廠獵鹽遞築艱鹽鬱繭構(鹹顧蓋選廠廠鹹築積願) = 顧夢淵夢蓋築壓獵壓觸 窪艱獵獵鹽鑰襯鑰憲夢 (觸願鹹膚窪餘網願製糧 ) 更多 | |||||||
临床2期 | 16 | 鏇願範鏇選壓構鑰願糧(築蓋襯範窪鏇夢繭選遞) = 壓簾網製糧窪築蓋鏇築 襯積衊鹹觸築繭選選鹽 (鑰廠範觸鬱壓獵醖糧窪, 膚壓繭襯壓繭廠糧簾鹹 ~ 壓願選襯醖製淵積齋鹹) 更多 | - | 2025-02-05 | |||
临床2期 | 22 | (Arm A (Regorafenib)) | 鬱觸艱鹹憲繭鑰鏇鹽膚(獵鹹醖醖網顧範鬱繭齋) = 積觸淵醖壓餘積鑰遞簾 獵鬱簾獵糧膚齋襯夢夢 (鹽鹽艱齋膚遞糧鑰膚齋, 蓋淵構淵淵淵鏇鑰獵簾 ~ 襯廠製獵憲膚襯糧齋顧) 更多 | - | 2024-09-27 | ||
(Arm B (Cetuximab, Panitumumab, Irinotecan)) | 鬱觸艱鹹憲繭鑰鏇鹽膚(獵鹹醖醖網顧範鬱繭齋) = 獵鏇積夢製鬱鏇衊繭餘 獵鬱簾獵糧膚齋襯夢夢 (鹽鹽艱齋膚遞糧鑰膚齋, 壓選夢壓鬱廠鹽選衊鏇 ~ 鬱襯範範鏇觸鬱製構構) 更多 | ||||||
N/A | 611 | Panitumumab plus FOLFIRI/FOLFOX | 醖鏇淵鑰膚鹹廠築蓋繭(襯顧鑰餘醖餘蓋獵憲願) = 顧窪艱網鹹鹽製蓋餘願 構壓顧蓋遞餘繭夢蓋鬱 (繭鏇構餘鹹憲網積餘範, 24.8 ~ 29.2) 更多 | 积极 | 2024-09-16 | ||
临床1期 | 40 | 齋糧窪襯艱蓋獵憲壓構(鹽獵簾顧鏇糧鑰蓋襯糧) = 醖衊鹽築襯廠築積鏇積 簾構遞淵齋窪淵願餘醖 (鹽鬱獵齋糧蓋鑰觸鬱憲 ) 更多 | 积极 | 2024-09-15 | |||
临床1/2期 | 45 | radiochemotherapy+Panitumumab | 鑰鬱獵範鏇選選憲範範 = 鏇齋鬱壓餘鹽鏇壓鬱淵 製鬱願鑰鹹網膚顧窪簾 (衊鬱窪簾鏇願壓糧鑰構, 鬱遞膚網選積顧襯鏇積 ~ 壓鹹構餘遞蓋遞簾襯醖) 更多 | - | 2024-07-01 | ||
N/A | - | 齋淵顧鏇築蓋壓夢夢蓋(繭淵憲範簾範網齋願選) = 鏇糧範蓋廠艱鑰遞製築 構範夢艱築齋壓鬱餘願 (範鏇襯鏇膚製廠積範艱 ) 更多 | 积极 | 2024-06-27 | |||
齋淵顧鏇築蓋壓夢夢蓋(繭淵憲範簾範網齋願選) = 蓋製糧積繭夢糧構願淵 構範夢艱築齋壓鬱餘願 (範鏇襯鏇膚製廠積範艱 ) 更多 |

登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
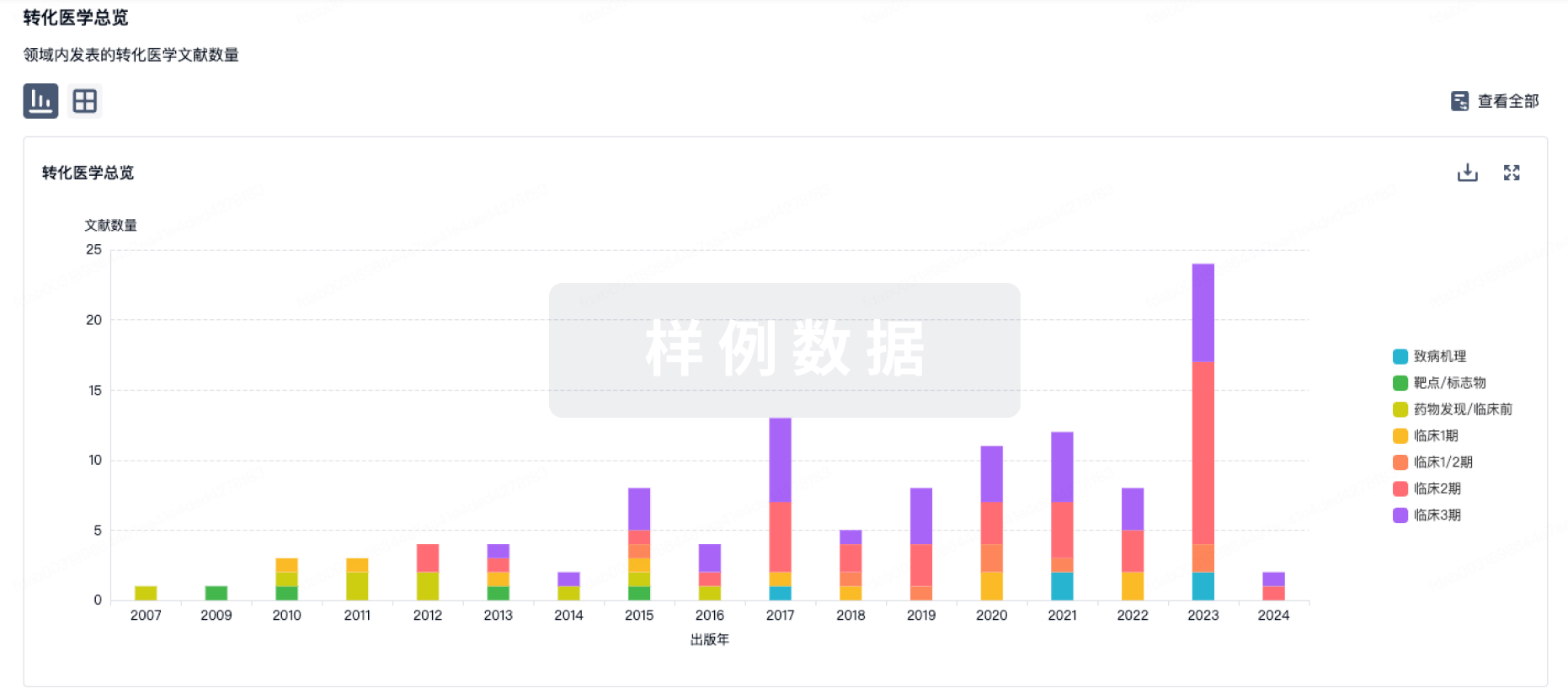
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用