PI3K靶点药物的机遇与挑战
免疫疗法蛋白降解靶向嵌合体抗体小分子药物快速通道
PI3K通过PI3K-AKT-MTOR通路来影响一系列的下游通道。磷脂酰肌醇-3-激酶(PI3K)/AKT/哺乳动物雷帕霉素(mTOR)信号传导靶标是最重要的细胞内途径之一,可调节细胞生长、存活、代谢和血管生成。
1、 肿瘤抑制因子PTEN的丧失或失活
2、 PI3K的突变或扩增
在癌症中最常见的突变或扩增是编码p110α亚基的基因,PI3KCA(磷脂酰肌醇3-激酶,催化,α多肽)。突变主要有以下几种类型:螺旋磷脂酰肌醇激酶同源结构域中的E545K(外显子9)突变,其降低调节亚基p85对p110α的抑制。H1047(外显子20)突变,H1047靠近催化结构域的末端,这增加了p110α与脂质膜的相互作用。E542K突变。
3、 PI3K上游酪氨酸激酶生长因子受体(如HER2,IGF-1),细胞粘附分子(如整合素,GPCR)或癌基因的激活。
全球药物研发现状
目前全球共研发了300多款PI3K药物,仅有5款药物上市,2款提交上市申请,59款处于临床阶段,65款处于临床前研究,23款处于药物发现阶段,138款终止研究或无研究进展。
已上市或提交上市申请的PI3K药物
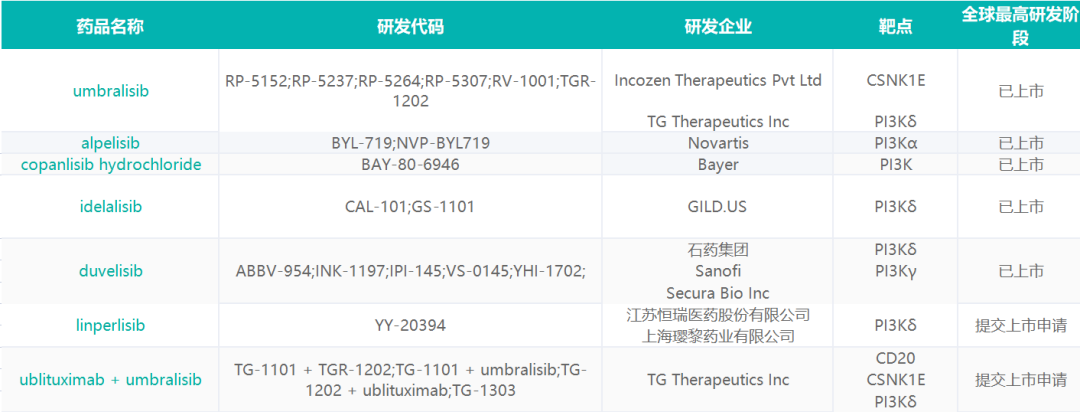
Preview
来源: 药通社
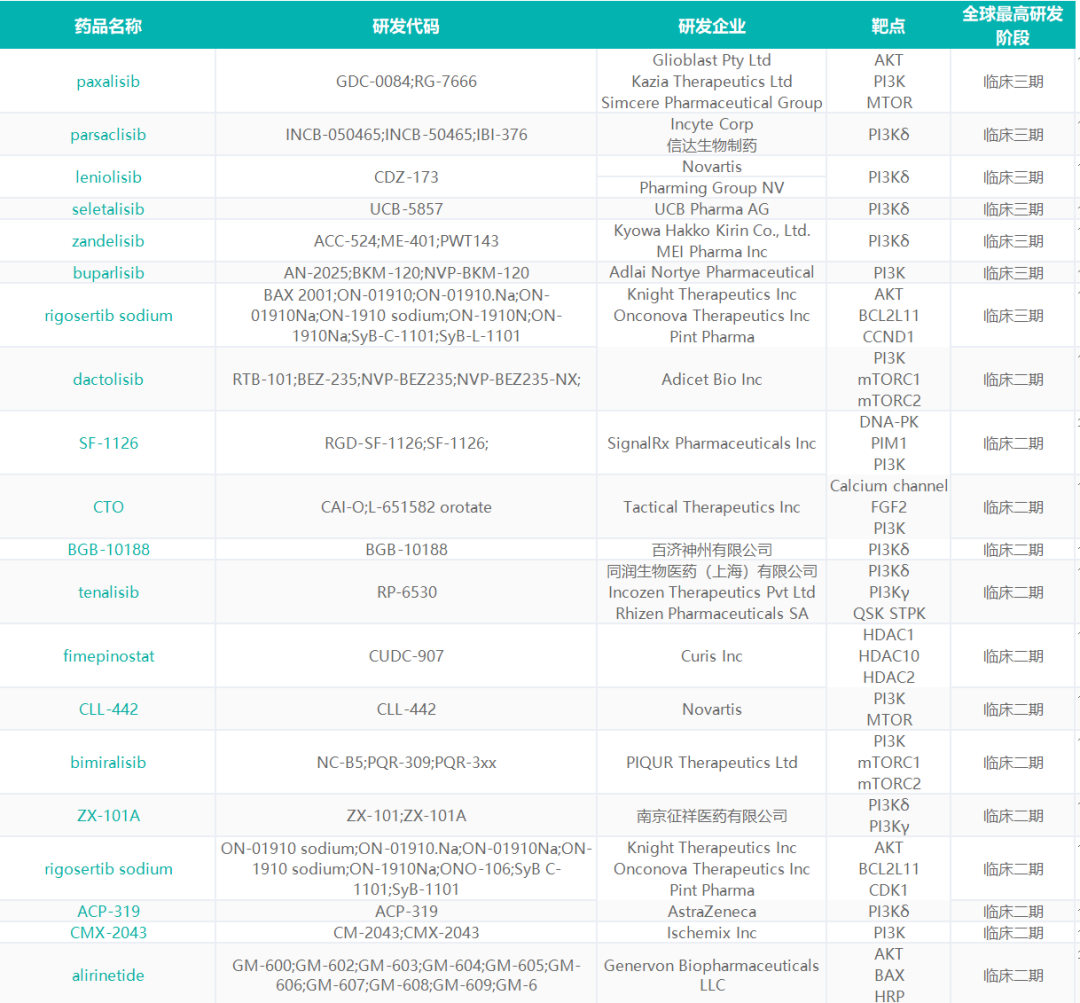
Preview
来源: 药通社
PI3K抑制剂
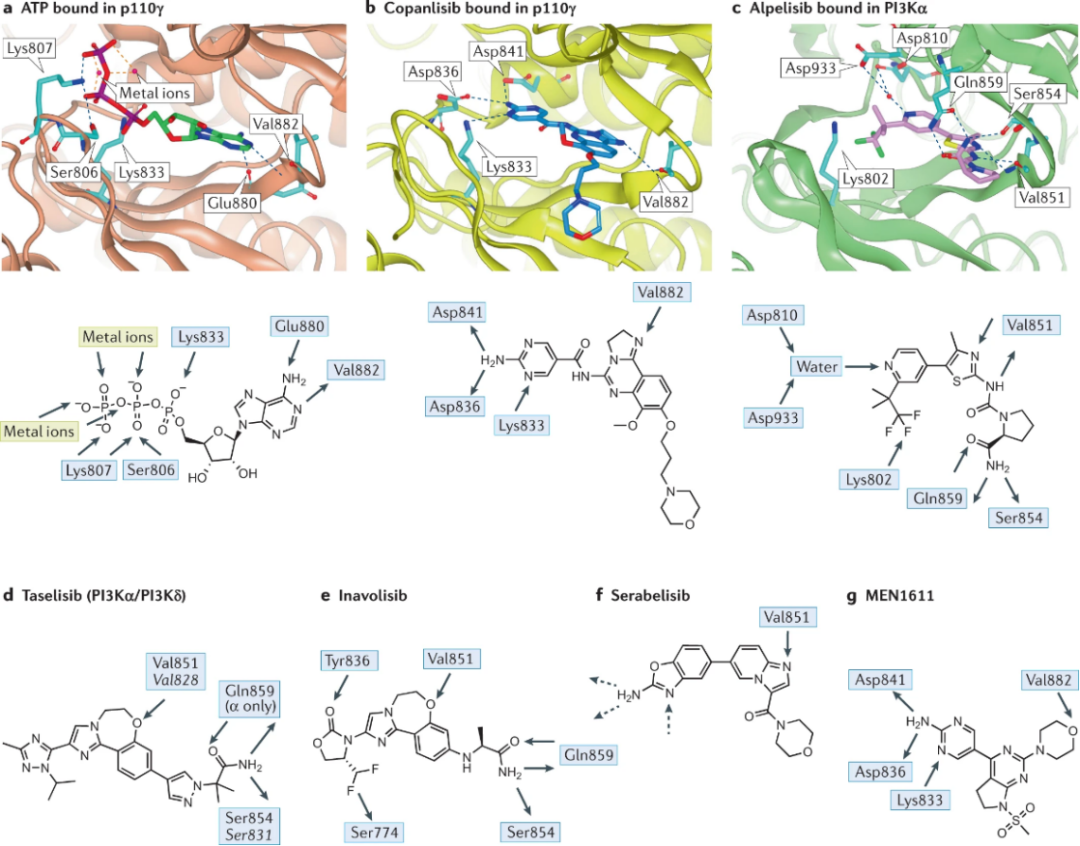
Preview
来源: 药通社
PI3K抑制剂按功能分为以下几种:
2017年,静脉注射的泛I类PI3K抑制剂copanlisib(拜耳)被批准用于复发性FL的成年患者。
2、异构体特异性PI3K抑制剂:异构体特异性PI3K 抑制剂是选择性抑制 p110α、p110β、p110δ 或 p110γ 催化亚基的药物。即PI3Kα抑制剂、PI3Kδ抑制剂、PI3Kγ抑制剂;相比泛PI3K抑制剂。高选择性的抑制剂的脱靶毒性更小,因此受到更多公司的青睐。
PI3Kα抑制剂
PI3Kδ抑制剂
Idelalisib是第一款PI3Kδ抑制剂,于 2014 年获批用于治疗 CLL、复发性 FL 和复发性小淋巴细胞淋巴瘤(SLL)。
Idelalisib单药的加速批准是基于一个单臂、多中心、开放标签的积极II期临床结果。该临床实验招募了123位复发性的“惰性”非霍奇金淋巴瘤(iNHL)和小淋巴细胞淋巴瘤(SLL)患者。病人每天接受两次,每次150毫克idelalisib治疗,一级实验终点是总应答率(ORR),二级实验终点是应答时间和无进展生存期。其中FL和SLL患者的总应答率分别为54%和58%。后者应答时间的中位数为11.9个月。这个结果和标准疗法的通常疗效相比相当或更好。
Linperlisib是江苏恒瑞医药股份有限公司和上海璎黎药业有限公司联合研发的一款PI3Kδ抑制剂。
在Linperlisib与Gemox联合治疗复发和/或难治性弥漫性大B细胞淋巴瘤的单臂1b/2期试验中,Linperlisib与GEMOX联合使用耐受性良好。总体反应率为60%(21/35),其中5人达到完全反应(CR,14%),16人达到部分反应(PR,46%),疾病控制率为71%(DCR),中位反应时间为36天。中位无进展生存期为193天,未达到中位反应持续时间,研究中仍有17个可评估点(48.6%)。治疗相关不良事件(TRAE)发生率为39%,其中大多数是1级或2级(83.9%)。不良反应发生率较低。
3、PI3K双亚型抑制剂:PI3Kδ/α抑制剂、PI3Kδ/γ抑制剂、PI3Kα/β
I期临床中,惰性非霍奇金淋巴瘤(iNHL)总体反应率,58%(n = 31)和6个完全反应(CR);复发/难治性 慢性淋巴细胞白血病(CLL), 56% (n = 55), 1 CR;外周T细胞淋巴瘤(TCL),50%(n = 16),3 CR;和皮肤TCL,32%(n = 19)。中位反应时间为∼1.8个月。84%的患者发生了严重(≥3级)不良事件。
4、PI3K多靶点抑制剂
Umbralisib是一种双 PI3Kδ/CK1ε 抑制剂。2020年,FDA授予umbralisib(PI3Kδ,CSNK1E靶点)与用于CLL的研究性CD20定向单克隆抗体ublituximabCD20定向单克隆抗体ublituximab联合使用的快速通道指定,并且在2021年,FDA批准了umbralisib用于边缘区(MZ)淋巴瘤和FL的加速批准。
与idelalisib和duvelisib相比, umbralisib的不良反应率更低,可能有以下几方面原因:(1) 高选择性:相对PI3Kα和PI3Kβ,umbralisib对PI3Kδ的选择性更高,比PI3Kγ具有大约225倍的选择性;(2)与大多数其他激酶抑制剂不同,umbralisib不通过经典的细胞色素P450 3A4(CYP3A4)代谢途径代谢,毒性更低。(3)耐受度提高: 通过抑制CK1-ε(酪蛋白激酶1ε),防止扰乱调节性T细胞的功能,从而提高药物的耐受性。PI3K inhibitors are finally coming of age。
PI3K药物新策略
PI3K降解剂
改善耐药策略
1、 结合PI3Kα正立构和变构药物变构药物
可以通过改变 ATP 结合位点并使其重新敏感来克服 PI3Kα 耐药性,即使其可接近空间阻断的 ATP 竞争性正构药物,或通过产生改变的、可成药的 PIP2 结合位点。
2、 抢救突变位置的变构药物
变构药物可以构成“锚”和“司机”。锚对接到一个变构口袋中,该口袋在非活动状态和活动状态之间的过渡期间构象保持不变。驱动因素“拉动”和/或“推动”蛋白质原子(侧链或骨架),并触发受体群体向药物结合有利的状态转移。
药物联用扩大治疗范围
口服可用的 parsaclisib (Incyte) 正在针对 B 细胞恶性肿瘤进行多项试验,并与抗 PD1 抗体联合用于一系列晚期实体瘤适应症 (NCT02646748, NCT03589651)目前已经完成试验,但还未透露试验结果。
小结
参考来源:
[1] PI3K inhibitors are finally coming of age.
[2] Development of PI3K inhibitors: Advances in clinical trials and new strategies (Review) | com.
[3] PI3K inhibitors: review and new strategies.
[4] Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients With Relapsed or Refractory Indolent Lymphoma.
[5] Targeting PI3K in cancer: mechanisms and advances in clinical trials.
[7] PI3K 选择性抑制剂作用机制的分子模拟研究.
内容来源于网络,如有侵权,请联系删除,谢谢。
来和芽仔聊天吧



立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。