预约演示
更新于:2025-08-29
Shanghai JMT Biological Technology Co Ltd
更新于:2025-08-29
概览
标签
肿瘤
呼吸系统疾病
消化系统疾病
单克隆抗体
小分子化药
抗体融合蛋白
疾病领域得分
一眼洞穿机构专注的疾病领域

暂无数据
技术平台
公司药物应用最多的技术
暂无数据
靶点
公司最常开发的靶点
暂无数据
| 排名前五的药物类型 | 数量 |
|---|---|
| 单克隆抗体 | 7 |
| 小分子化药 | 4 |
| 融合蛋白 | 2 |
| 抗体融合蛋白 | 2 |
| 脂质体药物 | 2 |
关联
17
项与 上海津曼特生物科技有限公司 相关的药物靶点 |
作用机制 PD-1抑制剂 |
在研适应症 |
最高研发阶段批准上市 |
首次获批国家/地区 中国 |
首次获批日期2024-06-25 |
靶点 |
作用机制 TOP1抑制剂 |
在研机构 |
原研机构 |
最高研发阶段批准上市 |
首次获批国家/地区 中国 |
首次获批日期2023-09-15 |
靶点 |
作用机制 RANKL抑制剂 |
在研机构 |
原研机构 |
最高研发阶段批准上市 |
首次获批国家/地区 中国 |
首次获批日期2023-09-05 |
50
项与 上海津曼特生物科技有限公司 相关的临床试验NCT07134205
A Randomized, Open-label, Multicenter Phase III Clinical Study of JMT101 in Combination With Irinotecan in Third-line and Beyond Treatment of Metastatic Colorectal Cancer
This is a randomized, open-label, multicenter, phase III clinical study. The aim is to evaluate the efficacy of JMT101 in combination with irinotecan in the third-line and beyond treatment of Metastatic Colorectal Cancer.
开始日期2025-10-01 |
申办/合作机构 |
NCT07140809
a Single-center, Randomized, Double-blind, Placebo-controlled, Multiple-dose Escalation Phase Ib/Ⅱa Study to Evaluate the Safety, Tolerability, Efficacy and Pharmacokinetic Characteristics of JMT202 Injection in Chinese Participants With Hypertriglyceridemia
To evaluate the safety and tolerability of multiple subcutaneous injections of JMT202 injection in Chinese participants with hypertriglyceridemia
开始日期2025-09-09 |
申办/合作机构 |
NCT07029139
A Multicenter, Randomized, Controlled, Open-label Phase II Clinical Study Evaluating the Efficacy and Safety of JMT601 Injection in Participants With Primary Membranous Nephropathy
This study is a multicenter, randomized, controlled, open-label, Phase Ⅱ clinical study to evaluate the efficacy, safety, Pharmacokinetics characteristics, Pharmacodynamics effects, and immunogenicity of JMT601 in participants with primary membranous nephropathy.
The study has two parts. Part one is dose escalation part, and Part two is dose expansion part.
The study has two parts. Part one is dose escalation part, and Part two is dose expansion part.
开始日期2025-06-30 |
申办/合作机构 |
100 项与 上海津曼特生物科技有限公司 相关的临床结果
登录后查看更多信息
0 项与 上海津曼特生物科技有限公司 相关的专利(医药)
登录后查看更多信息
170
项与 上海津曼特生物科技有限公司 相关的新闻(医药)2025-08-28
·贝壳社
作者:贝壳社
中国创新药行业已告别“野蛮生长”与“资本催熟”时期,步入理性竞争更为激烈的全新阶段。康宁杰瑞正是在这个新周期里一个具有观察价值的样本。
1
差异化战略
近年来,中国本土的药物研发能力取得了长足的进步,一批具有全球竞争力的首创新药成功获批上市。
例如,康方生物的PD-1/VEGF双特异性抗体依沃西单抗,是全球首个获批的同时靶向PD-1和VEGF两个关键通路的新型双特异性抗体,用于治疗非小细胞肺癌,兼具免疫治疗和抗血管生成作用。荣昌生物的泰它西普,是全球首个靶向B淋巴细胞刺激因子(BLyS)和增殖诱导配体(APRIL)的双靶点生物新药,用于治疗系统性红斑狼疮。这些药物不仅填补了临床空白,也标志着中国医药创新进入了新的发展阶段。
要理解康宁杰瑞,首先要从它在PD-1“百团大战”中的站位说起。面对PD-1领域的激烈竞争,康宁杰瑞选择避开锋芒,探索新的路径。
KN035的推出,彰显了康宁杰瑞对市场需求的精准把握。它没有去硬碰“K药”“O药”等,而是直击临床应用的最大痛点——给药方式。2021年11月,恩沃利单抗注射液(KN035),作为全球首个获批上市的皮下注射PD-L1抑制剂,正式在中国获得批准用于治疗不可切除或转移性微卫星高度不稳定性(MSI-H)或错配修复功能缺陷(dMMR)的成人晚期实体瘤患者。该药物的上市标志着一种新的治疗方式,能够为患者提供更为便捷的治疗选择。
对于需要长期治疗的患者而言,“便利性”本身就是一种极高的临床价值。这步棋,下的不是技术深度,而是商业智慧。
康宁杰瑞的估值与行业期待,则由其具有想象空间的‘双子星’产品——KN046与KN026所支撑。这是它试图构建的更深的技术护城河。
2
在“神仙打架”的赛道上寻找破局点
近日,安尼妥单抗注射液(KN026)已被国家药监局药审中心(CDE)纳入拟优先审评名单。拟适用于联合化疗治疗至少接受过一种系统性治疗(必须包含曲妥珠单抗联合化疗)失败的HER2阳性局部晚期、复发或转移性胃/胃-食管结合部腺癌。
KN026是康宁杰瑞开发的HER2双特异性抗体,可同时结合HER2的两个非重叠表位,阻断HER2信号。相比于曲妥珠单抗和帕妥珠单抗联用,KN026针对HER2阳性肿瘤细胞有更强的抑制杀伤。
2021年8月,康宁杰瑞与石药集团全资附属公司津曼特生物就KN026签订了在中国内地的开发及商业化授权协议。津曼特生物获得KN026在中国内地(不包括香港、澳门及台湾地区)在乳腺癌、胃癌适应症上的排他性开发与独占性商业化许可权。
2025年5月,康宁杰瑞与石药集团共同宣布,KN026联合化疗二线及以上治疗HER2阳性胃癌(包括胃-食管结合部腺癌)的Ⅱ/Ⅲ期临床研究(研究编号:KN026-001)期中分析达到无进展生存期(PFS)主要终点。详细数据暂未公布。
与此同时,KN026用于一线HER2阳性乳腺癌、HER2阳性乳腺癌新辅助等适应症的多项注册临床研究正在进行中。
HER2阳性胃癌约占所有胃癌的15%~20%,是一个非常重要的细分市场。KN026 (HER2双抗)这款产品诞生的背景,是HER2靶点“神仙打架”的时代。前有罗氏的曲妥珠单抗(赫赛汀),后有DS-8201(Enhertu)以“革命性”的数据横空出世。这留给后续研发者的市场空间已变得极为有限。
Enhertu是一款由阿斯利康和第一三共开发的HER2靶向ADC药物。基于DESTINY-Gastric系列研究的优异数据,已在全球多个国家获批用于治疗既往接受过含曲妥珠单抗方案治疗的HER2阳性局部晚期或转移性胃癌患者。它的出现,显著改善了患者的缓解率和生存期,是KN026必须直面的标杆。
然而,当前面临的挑战在于Enhertu作为HER2靶向ADC药物,在胃癌、乳腺癌等适应症中展现出显著疗效,尤其在二线及后线治疗中确立了较高疗效标准。KN026未来必须在特定的细分人群中找到自己不可替代的位置,否则极易被淹没在更强效的疗法之下。
此外,百济神州、再鼎医药在HER2阳性胃癌领域均有所布局,围绕胃癌治疗的多个靶点和方向进行多元化研发。
针对一线HER2阳性转移性乳腺癌,罗氏的“赫赛汀(曲妥珠单抗)+帕捷特(帕妥珠单抗)”双药联用方案,通过临床研究充分证明了同时阻断HER2不同结构域的价值。KN026的巧妙之处在于其独特的双特异性抗体设计,通过同时结合HER2的两个非重叠表位,实现了对HER2信号传导路径的更全面封锁,从而在理论上展现出克服赫赛汀耐药性的潜力。
3
揭盲失败
再看另一款双抗KN046(PD-L1/CTLA-4),CTLA-4是个被验证过有效、但存在毒性太大的问题。KN046采用机制不同的CTLA-4与PD-L1单域抗体融合组成,多项临床研究显示其在激活T细胞、增强抗肿瘤作用方面具有显著潜力。
但KN046却经历两次揭盲失败。2023年5月,KN046针对晚期鳞状非小细胞肺癌的III期临床试验中,总生存期(OS)未达到统计学显著性差异,未能成功揭盲。2024年5月,KN046治疗晚期胰腺导管腺癌的III期临床试验中,OS结果未达到预设的统计学终点,再次揭盲失败。
这两次失败对康宁杰瑞的股价和市场信心造成了重大冲击,公司市值一度大幅缩水。
创新是一场极其昂贵的马拉松。多款后期临床试验同时推进,意味着每个季度的研发费用都是天文数字。恩沃利单抗的销售虽能带来一定的资金回流,但相较于整个研发管线的庞大需求,其作用犹如杯水车薪。公司的现金流还能支撑多久?这是悬在管理层头上的达摩克利斯之剑。
目前,出海火热当下,一家中国Biotech的创新如果没有得到海外大型药企的认可,其“含金量”或许要打个问号。这不仅是钱的问题,更是对技术平台和产品质量的第三方权威背书。康宁杰瑞的KN046和KN026虽已备受瞩目多年,但市场仍在翘首以盼一笔能够彰显其价值的重磅全球交易。
康宁杰瑞或许诠释了当前中国Biotech的集体处境:手握着希望,也背负着压力;仰望着星空,也必须脚踏实地。
小结:康宁杰瑞凭借前瞻性的技术布局和差异化的产品策略,成功在中国创新药的“上半场”突围。然而,行业的“下半场”——比拼的将是真正的临床价值、全球商业化能力和资本效率。这也将是康宁杰瑞在未来发展中面临的重要挑战。
往
期
推
荐
1
医健企业前沿动态
2
医健行业BD/出海/投融资洞察
_
_
_
*声明:本文仅用于分享,不构成任何投资建议,且非治疗方案推荐
参考来源官方媒体/网络新闻
关于贝壳社
贝壳社是国内领先的医健创新创业服务平台,构建了"产业空间+创业教育+投资孵化+资本运作"四位一体服务体系,为医健领域创业企业提供全周期赋能。通过深度挖掘高成长项目、精准匹配产业资源及全球化生态网络,覆盖企业从孵化培育到加速发展的全链路,重点提供包括空间落地、创业培训、资本运作及跨境资源链接服务。贝壳社以加速科学技术成果转化与产业升级为目标,致力于成为医健领域的孵化器、加速器、连接器。
2025-08-24
据 Insight 数据库统计,本周(8 月 17 日—8 月 23 日)全球共有 87 款创新药(含改良新)研发进度推进到了新阶段,其中 5 款获批上市,5 款申报上市,23 款启动临床,11 款申报临床。
下文,Insight 将分别摘取本周国内外部分重点项目进展做介绍。
境外创新药进展
境外部分,本周共有 27 款药物研发阶段推进,包括 4 款获批上市,4 款申报上市,3 款首次启动临床。
获批上市
据 Insight 数据库显示,本周共有 4 条新药/新适应症(不含类似药)在三大海外主要国家/地区(美国、EMA、日本)获批。
本周在三大海外主要国家/地区获批的新药/新适应症
截图来自:Insight 数据库网页版
1、Ionis:又一款 ASO 疗法在美国获批,治疗遗传性血管性水肿
当地时间 8 月 21 日,Ionis Pharmaceuticals 宣布,美国 FDA 已批准其 「first-in-class」RNA 靶向药物 DAWNZERA™(Donidalorsen)上市,用于预防成人和 12 岁及以上的儿童患者遗传性血管性水肿(HAE)发作。
本次获批也使 Donidalorsen 成为全球首个针对 HAE 的 RNA 靶向药物。 此前,FDA 已授予 Donidalorsen 孤儿药资格。
截图来自:Ionis 官网
Donidalorsen 是 Ionis 开发的一款 RNA 靶向药物,靶向激肽释放酶原 (PKK) 。大冢制药拥有该产品在欧洲和包括日本在内亚太地区的独家商业化权利。欧洲药品管理局(EMA)也已接受该产品的上市许可申请,用于常规预防 12 岁及以上成人和青少年 HAE 的反复发作。
Donidalorsen 在美国的新药上市申请得到了关键性 Ⅲ 期 OASIS-HAE 研究,以及 Ⅱ 期 OLE 研究结果的支持。这些数据显示,Donidalorsen 可显著且持续地减少 HAE 发作,改善患者生活质量,且耐受性良好,大多数不良事件(AE)为轻度或中度。
其中, Ⅲ 期 OASIS-HAE 研究达到了研究的主要终点,与安慰剂相比,Donidalorsen 每四周一次治疗(Q4W),在 24 周内将患者月均 HAE 发作率降低 81%,关键次要终点显示,从第二次给药开始评估,月均 HAE 发作率显著降低 87%。此外,从第二次给药开始评估,Donidalorsen Q4W 在 24 周内将中度至重度 HAE 发作减少了约 90%。
该研究中,青少年患者在 0-24 周期间的 HAE 发作率较基线相比,Donidalorsen Q4W 组降低了 97%,而 Q8W 组降低了 71%。青少年患者 HAE 发作率的改善与 OASIS-HAE 研究的总体疗效结果一致。青少年患者报告了具有可接受的安全性特征和临床意义的生活质量改善,并且在治疗第 24 周时,所有患者均报告疾病控制良好。
Ionis 是反义寡核苷酸(ASO)领域的领军企业。Insight 数据库显示,截止目前,该公司已有 7 款 ASO 疗法获批上市(已退市药物除外),是获批 ASO 疗法最多的企业。
2、Madrigal:MASH 疗法在欧盟获批上市
当地时间 8 月 19 日,Madrigal Pharmaceuticals 宣布,欧盟委员会(EC)已有条件批准 Rezdiffra (resmetirom),用于治疗患有中度至晚期肝纤维化的非肝硬化性代谢功能障碍相关脂肪性肝炎(MASH)成人患者。
获批之后,Rezdiffra 成为欧盟(EU)首个也是目前唯一获批用于治疗 MASH 的疗法。
截图来源:GlobeNewswire
Rezdiffra 是一种每日口服一次、针对肝脏的甲状腺激素受体 β(THR-β)激动剂,旨在针对 MASH 的关键潜在病因进行治疗。
欧盟委员会的批准决定基于关键 3 期临床试验 MAESTRO - NASH 的积极结果,该试验达到了肝纤维化改善和 MASH 缓解的主要终点:1)与安慰剂组相比(10%),接受 80 mg 和 100 mg Resmetirom 治疗的患者中,26% 和 30% 实现了 MASH 症状缓解;2)与安慰剂组相比(14%),24% 和 26% 实现了至少一个阶段的纤维化改善,且非酒精性脂肪性肝病活动评分无恶化。
在 MAESTRO - NASH 试验中,Rezdiffra 还降低了肝脏硬度、肝脏脂肪含量、肝酶水平和致动脉粥样硬化脂质水平,并改善了与健康相关的生活质量。治疗一年后,使用 100mg Rezdiffra 治疗的患者中,有 91% 通过振动控制瞬时弹性成像(VCTE)检测显示肝脏硬度得到改善或保持稳定,这种检测方法在临床实践中常用于监测治疗反应。
MASH 领域是个市场空间巨大的蓝海赛道。据 Frost&Sullivan 预计,2023-2030 年,全球 MASH 患病人数将由 3.86 亿人增长至 4.86 亿人;中国 MASH 患病人数将由 0.43 亿人增长至 0.56 亿人;预计到 2030 年,全球 MASH 市场将达到 322 亿美元,复合年增长率为 24.6%,而我国将以 61.4% 的复合年增长率达到 355 亿元。
3、百济神州:重磅炸弹「泽布替尼」在欧洲获批片剂剂型
8 月 22 日,百济神州宣布,欧盟委员会(EC)已批准百悦泽®(泽布替尼)新的薄膜包衣片剂剂型用于所有当地已获批适应症。
截图来源:百济神州官方
泽布替尼片剂剂型旨在减轻患者的吞服负担、提升给药的便利性,从而有助于简化治疗方案,更好地满足患者在接受治疗中的真实需求。泽布替尼在全球范围内已治疗超过 20 万例患者,也是截至目前欧洲获批适应症最广的 BTK 抑制剂。
泽布替尼片剂剂型将于 2025 年 10 月起在欧洲启用,取代原胶囊剂型。片剂剂型可提升给药灵活性,便于医生为患者提供不同剂量的给药方案。另外,片剂剂型带有薄膜包衣,且比胶囊剂型体积更小,更易吞服。
其他监管动态
1、再生元/拜耳:「阿柏西普 8 mg」两项申请被 FDA 延期
当地时间 8 月 20 日,再生元宣布,FDA 已将两份 EYLEA HD®(阿柏西普)注射液 8 毫克监管申请的目标行动日期延长至 2025 年第四季度。这两份申请包括:
一份 EYLEA HD 预充式注射器的化学、生产和控制 (CMC) 预先批准补充文件 (PAS);
以及一份用于治疗视网膜静脉阻塞 (RVO) 后黄斑水肿患者的补充生物制品许可申请 (sBLA),该申请的原本 PDUFA 日期为 8 月 19 日。
截图来源:再生元官网
FDA 方面介绍,延期的原因是自近期对第三方制造商完成检查以来提供的信息构成了对每份申请的重大修订,因此延长了审查期限。
再生元则表示,此次延迟是预料之中的,原因是源于 FDA 对这些监管申请的灌装公司 Catalent Indiana LLC(已于 2024 年 12 月被诺和诺德收购)进行一般现场检查时观察到的情况。
Catalent 已于 2025 年 8 月初提交了一份全面的回复,以回应 FDA 指出的这些观察结果。再生元在新闻稿中表示,一旦生产问题得到解决,FDA 将能够迅速对这些申请作出监管审批决定。
目前,EYLEA HD 仍可通过小瓶给药在美国销售。该药物获批用于治疗湿性年龄相关性黄斑变性 (wAMD) 和糖尿病性黄斑水肿 (DME) 患者,给药间隔为每 8 至 16 周;用于治疗糖尿病性视网膜病变 (DR) 患者,初始给药间隔为每月 3 次。
临床试验结果
当地时间 9 月 6 日至 9 日,2025 年世界肺癌大会(WCLC 2025)将在巴塞罗那召开。 8 月 13 日,常规摘要正文已经发布。在大会开启前后,Insight 正在逐步追踪最新临床数据,相应文章合集可订阅 👉 2025 WCLC
1、荣昌生物:「维迪西妥单抗」肺癌 Ⅱ 期数据公布
荣昌生物维迪西妥单抗联合其它药物治疗肺癌的 Ⅱ 期临床数据在本次 WCLC 会议上进行了展示。
截图来源:WCLC 官网
维迪西妥单抗是首个获批的国产 HER2 靶向 ADC ,此前已在国内获批 3 项适应症,涵盖胃癌、 尿路上皮癌、乳腺癌。此外, 维迪西妥单抗用于HER2 低表达乳腺癌二线治疗、联合特瑞普利单抗治疗 HER2 表达的局部晚期或转移性尿路上皮癌的上市申请也已获得受理。
本次 WCLC 上公布的是一项单臂 II 期临床试验 RESOLUTION(NCT06749860)的数据,研究旨在评估维迪西妥单抗联合替雷利珠单抗及贝伐珠单抗用于 HER2 突变/扩增/表达的局部晚期或转移性非小细胞肺癌的疗效和安全性。
共 28 名符合条件的患者将接受维迪西妥单抗(2.0 mg/kg)、替雷利珠单抗(200 mg)和贝伐珠单抗(7.5 mg/kg)治疗,每 3 周静脉注射一次,直至病情进展或出现不可接受的毒性。为了实现对肿瘤动态的无创实时监测,循环肿瘤DNA (ctDNA) 检测被纳入该试验的关键探索性组成部分。ctDNA 分析采用 PredicineCARE(一种覆盖152个癌症相关基因的靶向新一代测序 (NGS) 检测)和 PredicineEPIC(一种全基因组甲基化检测)进行。
从 2024 年 4 月至 2025 年 1 月,共入组 11 例患者,其中 9 例可评估疗效。在这些患者中,3 例(33.3%)达到部分缓解,6 例(66.6%)病情稳定。确认的客观缓解率(ORR)为 33.3%,中位缓解持续时间为 5.1 个月。
研究共分析了 9 例患者的 29 份血液样本,并使用 PredicineCARE 检测方法进行分析。在所有样本中,该检测方法检测到 167 个突变和 148 个拷贝数变异(CNV)。基线时共发现 56 个突变和 79 个CNV。基线时最常发生变异的基因(>50% 的患者)是 ERBB2、TP53和 AKT3。根据每个样本中检测到的突变等位基因频率估算肿瘤分数。9 名患者中,5 名在 C3D1 处观察到肿瘤分数降低,其余患者维持稳定水平。值得注意的是,6 名患者早在 C5D1 处就检测到肿瘤分数增加,这与临床进展一致且早于临床进展。甲基化分析目前正在进行中。
综上,研究认为:维迪西妥单抗+替雷利珠单抗+贝伐珠单抗联合疗法是 HER2 基因突变型非小细胞肺癌 (NSCLC) 的一种有前景的治疗策略,支持进一步研究。此外,ctDNA 分析能够动态监测肿瘤负荷并早期发现疾病进展,凸显了其作为实时治疗反应评估的非侵入性工具的潜力。
2、Viking:GLP-1R/GIPR 双重激动剂口服药公布 2 期减重临床结果
当地时间 8 月 19 日,Viking Therapeutics 公布了口服 GLP-1R/GIPR 双重激动剂 VK2735 减重二期临床最新数据。当前,该药正在开发口服和皮下注射两种剂型,用于潜在治疗肥胖症等多种代谢疾病。
来源:企业官网
针对口服给药的 2 期临床试验 VENTURE 成功达到了主要和次要终点,接受 VK2735 治疗的患者与安慰剂组相比,体重出现了具有统计学意义的下降。此外,研究显示,在为期 13 周的每日给药过程中,VK2735 治疗安全且耐受性良好,大多数治疗期间出现的不良事件(TEAE)被归类为轻度或中度。
具体而言,每日一次口服 VK2735 片剂的受试者在 13 周后平均体重出现了具有统计学意义的下降,与基线相比降幅最高达 12.2%。与安慰剂组相比,接受 VK2735 治疗的受试者平均体重也出现了具有统计学意义的下降,降幅最高达 10.9%,且在 13 周时未观察到体重减轻出现平台期。
在评估体重减轻至少达到 5% 和 10% 的受试者比例这一关键次要终点上,所有剂量大于 15mg 的 VK2735 组与安慰剂组相比也均显示出具有统计学意义的差异。高达 97% 的受试者体重减轻达到或超过 5%,而安慰剂组仅为 10%;高达 80% 的受试者体重减轻达到或超过 10%,而安慰剂组仅为 5%。
在安全性方面,与安慰剂组相比,接受 VK2735 治疗的受试者因不良事件导致的停药率较低且均衡。研究期间,13% 接受安慰剂治疗的受试者因不良事件停止治疗,而接受 VK2735 治疗的受试者这一比例为 20%。治疗中断的最常见原因是胃肠道相关不良事件。
资格认定
1、BMS/百利天恒:双抗 ADC 获 FDA 突破性疗法认定
8 月 18 日,百利天恒宣布,美国 FDA 已授予 Izalontamab Brengitecan(iza-bren)突破性疗法认定,用于治疗携带 EGFR 19 号外显子缺失或 21 号外显子 L858R 置换突变、且在 EGFR-TKI 及铂类化疗后进展的局部晚期或转移性非小细胞肺癌患者。
来源:百利天恒官微
Iza-bren 是一款潜在全球首创双抗 ADC,其载荷为拓扑异构酶 1 抑制剂,可同时靶向表皮生长因子受体和人表皮生长因子受体 3(EGFRxHER3)。Iza-bren 通过双重作用机制阻断 EGFR 和 HER3 向肿瘤细胞传递的信号,从而抑制其增殖与存活信号。此外,通过抗体介导的内吞作用,Iza-bren 释放的治疗性有效载荷可引发基因毒性应激,最终导致肿瘤细胞死亡。
2023 年 12 月,百利天恒与 BMS 达成合作,Iza-bren 在中国由百利天恒独家开发,在中国以外的地区由西雅图免疫与百时美施贵宝共同开发,总金额高达 84 亿美元,其中首付款 8 亿美元.
2、第一三共/默沙东:B7-H3 ADC 获 FDA 突破性疗法认定
8 月 18 日,第一三共和默沙东合作开发的 B7-H3 ADC Ifinatamab deruxtecan(I-DXd)获得美国 FDA 授予突破性疗法认定(BTD),用于治疗在铂类化疗时或化疗后出现疾病进展的广泛期小细胞肺癌成人患者。
这是 ifinatamab deruxtecan 获得的首个 BTD,也是自第一三共与默沙东合作启动以来获得的首个 BTD。
Ifinatamab deruxtecan 采用第一三共独有的 DXd ADC 技术设计,由人源化抗 B7-H3 IgG1 单克隆抗体通过可裂解四肽连接子与多个拓扑异构酶 I 抑制剂有效载荷(一种依喜替康衍生物,DXd)连接组成。该药由第一三共和默沙东合作开发,现已获美国 FDA、欧盟委员会、日本厚生劳动省,以及中国台湾食品药物管理署授予孤儿药资格,用于治疗 SCLC。
此次 FDA 突破性疗法认定基于 IDeate-Lung01 II 期试验的数据,并获得 IDeate-PanTumor01 I/II 期试验的数据支持。
IDeate-Lung01 是一项全球、多中心、随机、开放性 II 期研究,以评估 ifinatamab deruxtecan 治疗广泛期小细胞肺癌患者的安全性和疗效。IDeate-Lung01 研究的主要分析结果将在国际肺癌研究协会主办的 2025 年世界肺癌大会(#WCLC25)上以最新重磅研究口头报告形式公布。
IDeate-PanTumor01 是一项全球、多中心、首次人体、开放性 I/II 期临床试验,以评估 ifinatamab deruxtecan 在对标准治疗无反应或无法耐受,或尚无标准治疗方案的晚期/不可切除或转移性实体瘤患者中的安全性和疗效。
医药交易
据 Insight 数据库显示,本周(8 月 17 日 - 8 月 23 日)共发生 23 起交易事件。
1、荣昌生物:眼科新药 13 亿元授权参天中国
8 月 19 日,荣昌生物发布公告,与日本参天制药株式会社全资子公司参天中国达成协议,将公司具有自主知识产权的 RC28-E 注射液有偿许可给参天中国。
根据协议条款,参天中国将获得 RC28-E 在大中华区及韩国、泰国、越南、新加坡、菲律宾、印度尼西亚及马来西亚的独家开发、生产和商业化权利,而荣昌生物将保留 RC28-E 在上述区域以外的全球独家权益。
财务方面,荣昌生物将从参天中国取得 2.5 亿元人民币的不可退还且不可抵扣的首付款,以及最高可达 5.2 亿元人民币的开发及监管里程碑付款和最高可达5.25亿元人民币的销售里程碑付款。此外,荣昌生物还将根据授权地区的产品销售额收取高个位数至双位数百分比的梯度销售分成。
RC28-E 是由荣昌生物自主研发的针对眼部新生血管性疾病的 VEGF/FGF 双靶标融合蛋白药物,可同时阻断 VEGF 和 FGF 家族的血管生成因子,从而更有效地抑制血管异常生长。该产品采用玻璃体腔内注射给药方式,且有望延长其半衰期、减少给药频率、减轻患者不适。
2025 年 5 月 7 日,RC28-E 治疗糖尿病性黄斑水肿(DME)的 Ⅱ 期临床试验数据在 2025 年美国眼科与视觉研究协会年会(ARVO 2025)上以口头报告形式进行公布。结果显示,RC28-E 在提高 DME 患者最佳矫正视力(BCVA)、降低黄斑中心区视网膜厚度 (CST)以及有效缓解黄斑水肿方面均表现出色。
2023 年,荣昌生物先后启动了 RC28-E 注射液治疗湿性年龄相关性黄斑变性(wAMD)和治疗糖尿病黄斑水肿(DME)的 Ⅲ 期临床试验。
2、复宏汉霖:引进启德医药临床 III 期创新 HER2 ADC
8 月 19 日,复宏汉霖宣布,与启德医药科技(苏州)有限公司(简称启德医药)达成战略合作。
根据约定,复宏汉霖将获得由启德医药开发的创新 HER2 靶向 ADC GQ1005 在中国及特定海外国家和地区开发和独家商业化权益。目前,该药物处于 III 期临床研究阶段,拟用于治疗 HER2 阳性乳腺癌。
GQ1005 是启德医药基于独创的酶促定点偶联技术开发的靶向 HER2 的创新 ADC 药物,通过稳定可裂解的开环连接子将拓扑异构酶 I 抑制剂与抗 HER2 单抗偶联而成。该药物采用高透膜性拓扑异构酶抑制剂作为有效载荷,具有强效旁观者杀伤作用,同时结合独特、稳定的连接子设计降低全身毒性,实现疗效与安全性的平衡。
临床前研究显示,GQ1005 在多个肿瘤细胞系异种移植模型中展现出与德曲妥珠单抗相当的抗肿瘤活性,且安全性优势明显。
与此一致,2024 年美国癌症研究协会年会 (AACR 2024) 公布了 GQ1005 治疗 HER2 表达或突变晚期实体瘤的 I 期临床数据,结果显示,GQ1005 在2.0mg/kg ~ 8.4mg/kg 剂量范围内均表现出良好的耐受性和安全性,同时在乳腺癌、胃癌、肺癌等多种实体肿瘤患者中表现出优异的治疗效果。
目前,该药物 III 期临床试验正在推进,GQ1005 的疗效与安全性将进一步被验证,其有望为 HER2 阳性乳腺癌患者带来更多生存获益。
乳腺癌是复宏汉霖重点布局的核心治疗领域。公司已建立覆盖乳腺癌全病程、全分子亚型的多元化产品管线,通过自建商业化团队和携手海外合作伙伴,组建了覆盖全球的商业化网络,持续释放乳腺癌管线的商业价值。
药企财报
跨国药企财报刚刚告一段落,又迎来了国内药企财报季。本周,多个国内药企密集释放上半年「成绩单」。Insight 摘取部分向读者分享。
1、中国生物制药:创新产品收入 78 亿元,同比增长 27.2%
8 月 18 日,中国生物制药发布 2025 上半年业绩报告,报告期内:收入 175.7 亿元,同比增长 10.7%;创新产品收入 78.0 亿元,同比增长 27.2%,占到了公司总收入的 44.4%;研发投入 31.9 亿元,占总收入约 18.1%。
中国生物制药业务覆盖医药研发平台、智能化生产和强大销售体系全产业链。产品包括多种生物药和化学药,涵盖肿瘤、肝病/代谢、呼吸系统、外科/镇痛四大治疗领域处于优势地位。
其中,上半年抗肿瘤用药收入约人民币 66.9 亿元,占集团收入约 38.1%,较去年同期增长约 24.9%。中国生物制药预计肿瘤领域未来三年(2025- 2027 年)将有 6 个创新药和 10 个生物类似药或仿制药获批上市。
2、恒瑞医药:上半年营收 157.6 亿元,创新药占比超 60%
8 月 20 日,恒瑞医药发布 2025 年业绩报告。报告期内,公司实现营业收入 157.61 亿元,同比增长 15.88%;归属于上市公司股东的净利润 44.50 亿元,同比增长 29.67%;归属于上市公司股东的扣除非经常性损益的净利润 42.73 亿元,同比增长 22.43%。
恒瑞正持续加大创新力度,维持较高的研发投入,报告期内公司累计研发投入 38.71 亿元,其中费用化研发投入 32.28 亿元。
2025 年上半年公司创新药销售及许可收入 95.61 亿元,占公司营业收入比重60.66%,其中创新药销售收入 75.70 亿元。创新药对外许可作为公司常态化业务,其收入已成为公司营业收入的重要组成部分。报告期内,公司收到 MerckSharp & Dohme 2 亿美元以及 IDEAYA 7500 万美元的对外许可首付款,并确认为收入,进一步推动经营业绩指标增长。
3、迪哲医药:实现商业化盈利,上半年营收大幅增长 74%
8 月 22 日,迪哲医药(股票代码:688192.SH)发布 2025 半年报。报告期内,公司实现营业收入 3.55 亿元,较上年度同比增长 74%。
2025 年上半年,迪哲医药在商业化方面,持续取得突破性进展。在医保赋能的推动下,旗下舒沃哲®(通用名:舒沃替尼片)和高瑞哲®(通用名:戈利昔替尼胶囊),销售持续放量,保持高速增长。
自我造血能力不断增强的同时,公司净亏损持续缩窄,同比下降 12%。公司账上现金及现金等价物达 22.51 亿元,同比增长 172%。
随着增收减亏的持续推进,迪哲医药首度实现商业化盈利,公司营收已覆盖研发费用之外的成本(销售、管理费用),财务可持续性闭环已经初具雏形。
4、亚盛医药:奥雷巴替尼上半年销售额大涨 93%
8 月 21 日,亚盛医药公布 2025 上半年业绩报告。上半年,亚盛医药的收入为 2.34 亿元人民币,比去年同期(8.24 亿元)下降 71.6%。亚盛医药指出收入下降主要由于该公司去年的收入记入了金额为 6.78 亿元人民币的知识产权收入。
其中,得益于国家医保目录对该药物覆盖范围的扩大,核心产品耐立克(奥雷巴替尼)上半年销售收入同比增长 93% 至人民币 2.17 亿元。
其它财务方面:
研发开支,上半年为 5.29 亿元人民币,同比增长 19.0%。
销售和分销开支,上半年为 1.38 亿元人民币,同比增长 53.7%,增长主要源于耐立克®的商业化扩张和利生妥®的上市准备工作。
截至 2025 年 6 月 30 日,亚盛医药现金和银行存款余额为 16.61 亿元人民币,同比 31.7%,这一增长主要得益于 2025 年 1 月美国首次公开发行带来的 9.7 亿元人民币净收益。
国内创新药进展
本周国内共有 65 款创新药(含改良新)研发进度推进到了新阶段,其中 4 款获批上市,2 款申报上市,5 款首次启动 III 期临床,17 款首次启动 I 期临床,16 款首次获批临床。
获批上市
1、轩竹生物/先声药业:新一代 ALK 抑制剂国内获批上市
8 月 22 日,NMPA 官网显示,轩竹生物/先声药业 1 类新药「地罗阿克」获批上市,单药适用于未经过间变性淋巴瘤激酶(ALK)抑制剂治疗的 ALK 阳性的局部晚期或转移性非小细胞肺癌(NSCLC)患者的治疗。
截图来源:NMPA 官网
地罗阿克(XZP-3621)是轩竹生物开发的新一代 ALK 抑制剂,其与 ALK 激酶结构域内的 ATP 结合位点结合,有效抑制 ALK 蛋白自磷酸化并阻断下游标靶的磷酸化。
此次获批是基于地罗阿克 III 期临床试验(NCT05204628)的数据。这是一项比较地罗阿克与克唑替尼治疗 ALK 阳性晚期 NSCLC 患者有效性和安全性的多中心、随机、开放 III 期临床研究。研究的主要终点是研究者评估的 PFS。
截至 2024 年 1 月 8 日,具体数据显示,地罗阿克在未经治疗的 ALK 阳性晚期 NSCLC 患者中表现出显著的抗肿瘤效果,ORR 为 86.9%(vs 克唑替尼 81.2%),mPFS 尚未达到(vs 克唑替尼 12.94 个月)。此外,达希替尼能够将疾病进展或死亡风险降低 57.8%(HR=0.422)。
在脑转移患者中,相较于克唑替尼,地罗阿克表现出更高的颅内客观缓解率(IC-ORR:92.3% vs 11.1%),中位颅内缓解持续时间(IC-DoR)未达到(vs 3.55 个月),mPFS 也未达到(vs 9.23 个月),HR 为 0.317。
在此前的一项 II 期临床试验中,接受地罗阿克治疗的患者(先前 ALK 抑制剂治疗出现疾病进展或不耐受)ORR 为 18.4%,DCR 为 65.3%。
除了本次获批的适应症以外,轩竹生物还在探索地罗阿克用于 ALK 阳性 NSCLC 患者的术后辅助治疗,以进一步拓宽产品的临床及商业价值。
据 Insight 数据库统计,此前 NMPA 已批准 11 款 ALK 抑制剂上市。地罗阿克此次获批成为国内获批的第 12 款 ALK 抑制剂。
2、第一三共/阿斯利康:TROP2 ADC 国内获批上市
8 月 22 日,NMPA 官网显示,第一三共/阿斯利康联合开发的 TROP2 ADC 德达博妥单抗(Datopotamab deruxtecan,Dato-DXd)获批上市,用于治疗既往接受过内分泌治疗且在晚期疾病阶段接受过至少一线化疗的不可切除或转移性的 HR 阳性、HER2 阴性(IHC 0、IHC 1+ 或 IHC 2+/ISH-)乳腺癌成人患者。
截图来自:NMPA 官网
此次德达博妥单抗获批上市是基于关键性 III 期临床 TROPION-Breast 01 研究的数据。这是一项全球、随机、多中心、开放性 III 期研究,旨在评估 Dato-DXd 与研究者选择的化疗方案(艾立布林、卡培他滨、长春瑞滨或吉西他滨)在经研究者评估的既往已接受内分泌治疗后疾病进展或不适合内分泌治疗的和接受过至少一种全身治疗的无法手术或转移性 HR 阳性、HER2 低表达或 HER2 阴性(IHC0,IHC1+ 或 IHC2+/ISH-)乳腺癌患者中的疗效和安全性。
TROPION-Breast01 的双主要终点为 PFS(由 BICR 评估)以及 OS。关键次要终点包括 ORR、缓解持续时间、研究者评估的 PFS、疾病控制率和至首次后续治疗时间。
在 2023 ESMO 大会上公布的研究结果显示,在主要终点 PFS 方面,经 BICR 评估,与研究者所选化疗(ICC)相比,Dato-DXd 用于内分泌经治的 HR 阳性、HER2 阴性(IHC0,IHC1+或 IHC2+/IHC-)转移性乳腺癌患者,可将疾病进展或死亡风险显著降低 37%(HR = 0.63;95% CI: 0.52-0.76;p<0.0001)。
Dato-DXd 治疗组的中位 PFS 为 6.9 个月,而化疗组 为 4.9 个月。在不同的亚组中均观察到一致的 PFS 获益。此外,Dato-DXd 组的 ORR 为 36.4%,而化疗组为 22.9%。
在针对研究另一双主要终点 OS 的期中分析中,截至数据截止日期,Dato-DXd 也显示出优于化疗 OS 改善趋势(HR = 0.84;95% CI: 0.62-1.14)。研究目前正在进行中,将对 OS 进行进一步评估。
安全性方面,Dato-DXd 整体安全性良好,未发现新的安全性问题。Dato-DXd 治疗组和化疗组 3 级或以上治疗相关不良事件(TRAE)发生率分别为 21% 和 45%,Dato-DXd 组不到化疗组的一半。
2024 年 9 月,第一三共和阿斯利康共同宣布 Dato-DXd 的 III 期 TROPION-Breast01 研究未达到 OS 主要终点。
阿斯利康表示,患者的 PFS 获益未能成功转化为 OS 统计学意义上的改善,可能与该项临床试验进行期间患者所接受的后线治疗有关。包括 Enhertu 在内的 ADC 药物期间获批上市,参与试验的患者在进展后接受了这些药物治疗,影响了 OS 结果。
Insight 数据库显示,在乳腺癌领域,第一三共/阿斯利康共开展了 5 项 III 期临床试验,除针对 HR+/HER2- 乳腺癌外,还包括转移性三阴性乳腺癌一线疗法以及辅助疗法等。
在乳腺癌之外,同时也在推进在肺癌领域的进展。25 年 6 月,该药获 FDA 批准上市,用于治疗既往接受过 EGFR 靶向疗法和铂类化疗的局部晚期或转移性 EGFR 突变非小细胞肺癌 (NSCLC) 成人患者。
此外,两家公司也在探索 Dato-DXd 用于尿路上皮癌、结直肠癌、子宫内膜癌、胃癌等其他癌种的潜力。
3、诺华:IgA 肾病新药首次在国内获批上市
8 月 20 日,诺华宣布,诺华的阿曲生坦在中国获批上市,用于降低有疾病进展风险的原发性免疫球蛋白 A 肾病(IgAN)成人患者的蛋白尿(受理号:JXHS2400103)。该适应症此前已被 CDE 纳入优先审评。
新闻稿指出,这是国内首个获批治疗 IgA 肾病的非免疫性疗法,是国内首个且目前唯一针对该适应症的高选择性内皮素 A(ETA)受体拮抗剂,有望重塑 IgA 肾病治疗基石。
截图来源:NMPA 官网
阿曲生坦是一种在研的强效选择性口服 ETA 受体拮抗剂。2025 年 4 月,该药首次获 FDA 加速批准用于 IgA 肾病,成为首个也是唯一一个用于该适应症的强效选择性口服 ETA 受体拮抗剂。
2024 年 5 月,诺华公布了阿曲生坦治疗 IgA 肾病患者的 III 期 ALIGN 研究的预定中期分析结果。这是一项 III 期、随机、双盲、安慰剂对照研究,旨在比较阿曲生坦和安慰剂在存在进行性肾功能丧失风险的 IgA 肾病中的疗效和安全性。
结果显示,与接受支持治疗(最大耐受剂量和稳定剂量的肾素-血管紧张素系统 [RAS] 抑制剂)的患者相比,接受阿曲生坦治疗的患者在 36 周时蛋白尿(以 24 小时尿蛋白与肌酐比率 [UPCR] 衡量)减少了 36.1% (p<0.0001)。研究还显示,阿曲生坦具有良好的安全性,与前期报道的数据一致。
Insight 数据库显示,在 IgA 肾病领域,除了本次获批上市的阿曲生坦以外,诺华还有 3 款产品,旨在针对不同的 IgA 肾病驱动因素,为患者群体提供多样化的治疗选择。
申报上市
1、恩华药业:麻醉 1 类新药报上市
8 月 20 日,CDE 官网显示,恩华药业 NH600001 乳状注射液申报上市。根据公开资料和临床试验进展,Insight 数据库推测适应症为用于短时手术麻醉。
截图来源:CDE 官网
NH600001 分子结构与依托咪酯相似,拟开发成与依托咪酯相比具有明显优势的短效静脉麻醉 1 类新药。
临床前研究结果表明,NH600001 乳状注射液保留了依托咪酯麻醉起效快、苏醒迅速、安全窗大、对呼吸和循环系统影响小等优点。同时克服了依托咪酯抑制肾上腺皮质功能的缺点,对肾上腺皮质激素没有明显的抑制作用。
2024 年 5 月,恩华药业曾启动一项多中心、随机、双盲、阳性药(依托咪酯)平行对照的 III 期临床研究,旨在评价 NH600001 乳状注射液用于胃镜和结肠镜诊疗镇静/麻醉的有效性和安全性。该试验于 24 年 9 月完成,目前结果尚未披露。
2、阿斯利康:红斑狼疮新药国内报上市
8 月 20 日,CDE 官网显示,阿斯利康阿伏利尤单抗注射液报上市,用于治疗系统性红斑狼疮(SLE)。
截图来源:CDE 官网
阿伏利尤单抗是全球首个获批的靶向 Ⅰ 型干扰素通路的 SLE 治疗靶向药物。研究显示,阿伏利尤单抗能够改善患者临床表现,降低疾病活动度,减少疾病复发,帮助患者实现更好的疾病预后。
该药于 2021 年 7 月获得 FDA 批准,用于治疗中度至重度 SLE 成人患者,并于 2021 年 9 月、2022 年 2 月先后在日本和欧盟获批上市。自首次获批以来,阿伏利尤单抗销售额一路上涨,24 年全球销售额达 4.74 亿美元,同比大增 69.29%。
3、强生:复方新药新适应症国内报上市
8 月 22 日,CDE 官网显示,强生尼拉帕利阿比特龙片新适应症上市申请获受理。根据公开资料和临床进展,Insight 数据库推断本次申报上市的适应症为激素依赖性前列腺癌。
截图来源:CDE 官网
尼拉帕利阿比特龙是强生推出的复方制剂,于 2023 年先后在欧盟、美国获批上市,用于联合泼尼松或泼尼松龙治疗 BRCA1/2 突变的转移性去势抵抗性前列腺癌成人患者(mCRPC)一线治疗。2024 年 10 月,该药首次在国内获批上市,成为国内首个且唯一获批的双效复方制剂。
2025 年 7 月,强生宣布向 EMA 提交了尼拉帕利阿比特龙扩展适应症的申请,寻求批准该药与泼尼松或泼尼松龙联合用于治疗激素依赖性前列腺癌,该申请得到了 AMPLITUDE 研究数据的支持。而 Insight 数据库推断,本次在国内报上市的适应症与此相同。
在 2025 年 ASCO 大会上,强生以口头报告的形式首次公布了 AMPLITUDE 研究的临床结果。这是一项 III 期、随机、安慰剂对照、双盲研究,旨在携带有害胚系或体系同源重组修复(HRR)基因突变的转移性去势敏感性前列腺癌(mCSPC)受试者中评估尼拉帕利阿比特龙联合泼尼松加雄激素剥夺疗法的疗效和安全性。
696 名患者随机分配至尼拉帕利阿比特龙+泼尼松组(试验组,n=348)或安慰剂+泼尼松组(对照组,n=348),55.6% 的患者存在 BRCA1/2 基因变异。主要终点是经研究者评估的 rPFS。
中位随访时间为 30.8 个月,该研究的主要终点达成,整体试验组的中位 rPFS 尚未达到,但显著长于对照组(29.5 个月)。在预设的 BRCA1/2 亚组中显示出相似的结果,试验组中位 rPFS 尚未达到,对照组中位 rPFS 为 26 个月。
在次要终点方面,试验组的症状进展时间(TSP)显著改善,症状进展风险降低了 50%,而在 BRCA1/2 亚组中症状进展风险降低了 56%。此外,在首次中期分析中,试验组具有改善总体 OS 的趋势。
安全性方面,3/4 级不良事件 (AE) 在试验组中发生率为 75.2%,在对照组中发生率为 58.9%,最常见的是贫血和高血压。因 AE 导致的停药的发生率较低(11.0% vs 6.9%)。
4、爱科百发:RSV 治疗药物再次申报上市
8 月 20 日,CDE 官网显示,爱科百发「齐瑞索韦肠溶胶囊」上市申请获受理,推测用于由呼吸道合胞病毒(RSV)引起的 2 岁及以下儿童呼吸道感染的治疗。
截图来源:CDE 官网
齐瑞索韦(Ziresovir,爱司韦®)是一款全新的靶向 RSV 融合蛋白小分子抑制剂,通过与病毒的 F 蛋白结合从而阻止病毒侵入人体细胞。该药是首个获国家药品监督管理局(NMPA)突破性治疗品种认定的非肿瘤创新药。
齐瑞索韦是全球首个成功完成 III 期临床试验并获得积极结果的靶向 RSV 的特效抗病毒药物,也是首个在中国发明和开发,并拓展到全球的儿童创新药。其 III 期临床结果已在《新英格兰医学杂志》和《柳叶刀·儿童和青少年健康》杂志发表。在 III 期临床试验中,齐瑞索韦显示出以下核心优势:
优异的安全性:在包括新生儿在内的儿童群体中未发现与治疗相关的严重不良反应,安全性良好;
强效抗病毒效应:用药 5 天后病毒载量较安慰剂组显著降低,加速感染病毒清除过程;
快速改善症状:患儿呼吸道症状严重程度评分显著下降,住院时间缩短, 临床获益显著;
长期健康获益:两年随访期复发性喘息和哮喘发生率降低,具有长期健康受益。
作为一款儿童用药,齐瑞索韦 10mg 肠溶微丸胶囊剂型完美兼顾疗效、安全性和使用便利性:
灵活给药:胶囊可打开,胶囊内药物可混合于酸奶、苹果酱或水服用,有效解决婴幼儿吞咽困难的问题;
剂量精准:10mg规格便于按体重灵活组合剂量,避免碎片化制剂问题,提升用药准确性;
适用场景广:口服剂型适用多种场景和不同年龄儿童及婴幼儿,尤其适合医疗资源匮乏地区。
此前于 2022 年 12 月,齐瑞索韦首次在国内申报上市获受理,不过于 2024 年 2 月收通知件暂未批准。本次再次申报上市,意味着国内儿科患者距离这一创新疗法又近一步。
拟优先审评
1、康宁杰瑞/石药集团:HER2 双抗拟纳入优先审评
8 月 20 日,CDE 官网显示,津曼特生物申报的安尼妥单抗注射液纳入拟优先审评,适应症为联合化疗用于至少接受过一种系统性治疗(必须包含曲妥珠单抗联合化疗)失败,HER2 阳性局部晚期、复发或转移性的胃/胃-食管结合部腺癌。
截图来源:CDE 官网
安尼妥单抗(KN026)是康宁杰瑞自主研发的 HER2 双特异性抗体,可同时结合 HER2 的两个非重叠表位,导致 HER2 信号阻断,优于曲妥珠单抗或者帕妥珠单抗单用,达到曲妥珠单抗和帕妥珠单抗联用的效果,如展示出更高的亲和力,在 HER2 阳性肿瘤细胞株中具备优效的肿瘤抑制作用。同时,KN026 对 HER2 中低表达肿瘤和曲妥珠单抗抗性细胞株也有抑制作用。
2021 年 8 月,康宁杰瑞与石药集团就 KN026 达成一项 10 亿元的独家授权许可协议,协议授权石药集团全资附属公司津曼特生物在中国内地(不包括香港、澳门或台湾)在乳腺癌、胃癌适应症上的排他性开发与独占性商业化许可权。
在 2022 年 ASCO 大会上,康宁杰瑞公布了 KN026 单药后线治疗晚期 HER2 表达的胃癌或胃食管结合部癌(GC/GEJ)的 II 期临床研究数据。
这是一项多中心、开放标签、双队列的 II 期研究,入组至少一线系统治疗后进展的晚期 GC/GEJ 患者,队列 1 为 HER2 高表达(IHC3+ 或 IHC 2+ /ISH+),队列 2 为 HER2 低表达(IHC 2+ /ISH-, IHC 1+ 或 IHC 0/ISH-)。主要终点是ORR 和 DOR。
截至 2021 年 10 月 29 日,共入组了 45 例患者,其中 39 例(队列 1 有 25 例,队列 2 有 14 例)患者可进行有效性评估:
在队列 1 中,ORR 为 56%,DOR为 9.7 个月,中位 PFS 为 8.3 个月,中位 OS 为 16.3 个月;
队列 1 中 14 例患者此前接受过曲妥珠单抗治疗,其 ORR 为 50%,中位 DOR 为 7.0 个月,中位 PFS 为 5.5 个月,中位 OS 为 14.9 个月。
在队列 2 中, ORR 为 14%,中位 DOR 为 6.2 个月,中位 OS 为 9.6 个月。
安全性方面,45 例入组患者全部纳入了安全性分析,在 37 例患者中观察到治疗相关的不良事件(TRAE)。最常见的 TRAE(任何级别)为天冬氨酸氨基转移酶升高、丙氨酸氨基转移酶升高、皮疹、贫血及输液相关反应。4 例患者报告了 5 次 3 级 TRAE,包括输液相关反应、肾鞘膜积液、输尿管狭窄、血压升高和肝功能异常。未发生 4 级或 5 级 TRAE。
2、艾伯维:重磅双抗拟纳入优先审评
8 月 21 日,CDE 官网显示,艾伯维递交的艾可瑞妥单抗注液拟纳入优先审评,适应症为联合利妥昔单抗和来那度胺适用于治疗复发或难治性滤泡性淋巴瘤(FL)成人患者。在针对这一适应症的 Ⅲ 期研究中,艾可瑞妥单联合疗法组的患者 ORR 为 95.7%。
截图来自:CDE 官网
艾可瑞妥单抗(Epcoritamab)是一款 CD20×CD3 双抗,最初由 Genmab 公司开发 。 2020 年,艾伯维与 Genmab 达成一项总额达 39 亿美元的合作,以共同开发和商业化 Genmab 的三种下一代双抗产品,其中就包括 Epcoritamab。
艾可瑞妥单抗于 2023 年 5 月全球首批。截至目前,该药已在美国、欧盟、日本获批上市,获批适应症涵盖弥漫性大 B 细胞淋巴瘤、滤泡性淋巴瘤、原发纵隔大 B 细胞淋巴瘤。2024 年,艾可瑞妥单抗的全球销售额为 2.81 亿美元。
在国内,艾伯维在 24 年 11 月递交了艾可瑞妥单抗注射用浓溶液的首个上市申请,推测适应症为复发/难治性弥漫性大 B 细胞淋巴瘤。本次拟优先审评针对的适应症为艾可瑞妥单抗联合利妥昔单抗和来那度胺适用于治疗复发或难治性滤泡性淋巴瘤。
在关键 Ⅲ 期研究 EPCORE FL-1(NCT05409066)试验中,研究人员评估了艾可瑞妥单抗+利妥昔单抗+来那度胺治疗复发/难治性滤泡性淋巴瘤患者的安全性和有效性,对照组为利妥昔单抗+来那度胺。试验的双重主要终点是 ORR 和 PFS。
该试验的首次中期分析数据显示,ORR 为 95.7%(p 值< 0.0001),PFS(HR 0.21,p 值< 0.0001)也具有显著统计学意义。基于这一结果,美国 FDA 已在今年 7 月 24 日受理艾可瑞妥单抗+利妥昔单抗+来那度胺治疗复发/难治性滤泡性淋巴瘤的上市申请,并授予其优先审评,PDUFA 日期为 2025 年 11 月 30 日。
2025 年 8 月 7 日,Genmab 宣布 EPCORE FL-1 临床试验取达到了 ORR 和 PFS 两个主要终点。艾可瑞妥单抗组在两个终点上均显示出具有统计学意义和临床意义的差异,将疾病进展或死亡风险降低了 79%。研究的具体数据将于第 67 届美国血液学会(ASH)年会上展示。
临床成功
1、康方生物:IL-17A 单抗又一适应症 Ⅲ 期临床成功
8 月 21 日,康方生物宣布,其自主研发的新型人源化抗 IL-17A 单克隆抗体古莫奇单抗(研发代号:AK111),在治疗活动性强直性脊柱炎(AS)的关键注册性 Ⅲ 期临床研究中取得阳性结果:主要疗效终点 ASAS20 及亚组分析、关键次要终点 ASAS40,及其他预先选定的多个次要终点均获得成功,具有统计学显著性和临床意义的改善。
截图来源:康方官微
数据显示,古莫奇单抗高效、快速缓解了患者 AS 症状,显著改善了患者疾病活动度的同时,也显著改善了患者的躯体功能及生活质量,有望为中国 AS 患者提供高效安全的治疗新选择。
古莫奇单抗是康方生物自主研发的一款靶向于人 IL-17A 的 IgG1 单克隆抗体,为生物抗体 I 类新药。目前,古莫奇单抗治疗中、重度斑块状银屑病、强直性脊柱炎的注册性 Ⅲ 期临床研究均已达到全部疗效终点。另外,古莫奇用于治疗中度至重度斑块型银屑病的新药上市中请已于 2025 年 1 月获得 CDE 受理。
古莫奇单抗是康方生物第 3 个提交上市申请的非肿瘤药物。随着伊努西单抗、依若奇单抗、古莫奇单抗、曼多奇单抗等产品相继上市,及 IL-4R/ST2 双抗、神经退行性病变等非肿瘤创新靶点药物针对多适应症深度开发的高效推进,康方生物非肿瘤板块的全球竞争力正在不断增强。
启动临床
1、应世生物:全球首创 FAK 抑制剂启动 III 期临床
8 月 19 日,药物临床试验登记与信息公示平台官网显示,应世生物登记了一项评估 IN10018(Ifebemtinib)联合 D-1553(格索雷塞)对比标准治疗在一线 KRAS G12C 突变阳性的局部晚期或转移性非鳞状非小细胞肺癌中的随机、对照、开放性、多中心 III 期研究(IN10018-023)。
这是 IN10018 启动的首个 III 期临床。
来源:药物临床试验登记与信息公示平台官网
该研究旨在评估 IN10018 联合 D-1553 对比标准治疗在一线 KRAS G12C 突变阳性的局部晚期或转移性非鳞状 NSCLC 受试者中的有效性和安全性,计划入组 400 名受试者,随机分组接受治疗。
主要终点是 BICR 根据 RECIST v1.1 标准评估的 PFS,次要终点包括 BICR 评估的 ORR、DCR 和 DoR 等,研究者评估的 ORR、DCR、DoR 和 PFS 等,OS 以及安全性指标。
IN10018 是一款靶向黏着斑激酶(FAK)的强效、口服、高选择性的全球首创小分子抑制剂,2019 年应世生物从 BI 引进。
D-1553 是益方生物和正大天晴合作开发的一款 KRAS G12C 抑制剂,用于治疗带有 KRAS G12C 突变的非小细胞肺癌、结直肠癌等多种癌症。2024 年 11 月,该产品已获国家药监局附条件批准上市,用于治疗至少接受过一种系统性治疗的 KRAS G12C突变型的晚期 NSCLC 成人患者。
2、齐鲁制药:又一款 1 类新药启动 III 期临床
8 月 21 日,药物临床试验登记与信息公示平台官网显示,齐鲁制药登记了一项注射用 QLS32015 用于复发/难治性多发性骨髓瘤(RRMM)的 III 期试验。
公开资料显示,这是该药启动的首个 III 期临床试验。
截图来源:药物临床试验登记与信息公示平台官网
这是一项随机、对照、多中心、开放的 III 期研究,旨在评估注射用 QLS32015 单药和研究者选择的方案在既往接受过 ≥3 线治疗的复发或难治性多发性骨髓瘤受试者中的无进展生存期(PFS)。
QLS32015 是齐鲁制药自主研发的一款靶向 GPRC5D 和 CD3 的新型人源化 IgG1 T 细胞重定向双特异性抗体,通过不依赖于主要组织相容性复合体和特异性 T 细胞受体结合的途径,形成免疫突触,激活 T 细胞,诱导 T 细胞对肿瘤细胞的杀伤作用。
在 2024 年美国血液学会(ASH)年会上,齐鲁公布首次人体试验结果。该研究是一项开放标签、剂量递增/扩展的 I 期临床试验,旨在探索 QLS32015 在 RRMM 患者中的安全性和有效性。截至 2024 年 8 月 31 日,共入组 13 例患者。至数据截止时间,13 例患者中有 12 例患者至少接受过一次疗效评估。根据国际骨髓瘤工作组(IMWG)标准评估的 ORR 为 76.9%,其中有 2 例患者达到完全缓解(CR),3 例患者达到非常好的部分缓解(VGPR),5 例患者达到部分缓解(PR)。
安全性方面,在 54 μg/kg 剂量水平发生 1 例 DLT,MTD 未达到。治疗相关不良事件(TRAEs)的发生率为 100%。最常见的 TRAEs 为细胞因子释放综合征,所有 CRS 均为 1 级或者 2 级,中位持续时间为 2.1 天。
内容来源:药企官方发布新闻/资料、Insight 数据库
封面来源:站酷海洛 Plus
免责声明:本文仅作信息分享,不代表 Insight 立场和观点,也不作治疗方案推荐和介绍。如有需求,请咨询和联系正规医疗机构。
PR 稿对接:微信 insightxb
投稿:微信 insightxb;邮箱 insight@dxy.cn
点击卡片进入 Insight 小程序
国内审评进度、全球新药开发…
随时随地查!
多样化功能、可溯源数据……
Insight 数据库网页版等你体验
点击阅读原文,立刻解锁!
临床3期上市批准财报孤儿药临床2期
2025-08-20
8 月 20 日,CDE 官网显示,津曼特生物申报的安尼妥单抗注射液纳入拟优先审评,适应症为联合化疗用于至少接受过一种系统性治疗(必须包含曲妥珠单抗联合化疗)失败,HER2 阳性局部晚期、复发或转移性的胃/胃-食管结合部腺癌。
截图来源:CDE 官网
安尼妥单抗(KN026)是康宁杰瑞自主研发的 HER2 双特异性抗体,可同时结合 HER2 的两个非重叠表位,导致 HER2 信号阻断,优于曲妥珠单抗或者帕妥珠单抗单用,达到曲妥珠单抗和帕妥珠单抗联用的效果,如展示出更高的亲和力,在 HER2 阳性肿瘤细胞株中具备优效的肿瘤抑制作用。同时,KN026 对 HER2 中低表达肿瘤和曲妥珠单抗抗性细胞株也有抑制作用。
2021 年 8 月,康宁杰瑞与石药集团就 KN026 达成一项 10 亿元的独家授权许可协议,协议授权石药集团全资附属公司津曼特生物在中国内地(不包括香港、澳门或台湾)在乳腺癌、胃癌适应症上的排他性开发与独占性商业化许可权。
在 2022 年 ASCO 大会上,康宁杰瑞公布了 KN026 单药后线治疗晚期 HER2 表达的胃癌或胃食管结合部癌(GC/GEJ)的 II 期临床研究数据。
这是一项多中心、开放标签、双队列的 II 期研究,入组至少一线系统治疗后进展的晚期 GC/GEJ 患者,队列 1 为 HER2 高表达(IHC3+ 或 IHC 2+ /ISH+),队列 2 为 HER2 低表达(IHC 2+ /ISH-, IHC 1+ 或 IHC 0/ISH-)。主要终点是ORR 和 DOR。
截至 2021 年 10 月 29 日,共入组了 45 例患者,其中 39 例(队列 1 有 25 例,队列 2 有 14 例)患者可进行有效性评估:
在队列 1 中,ORR 为 56%,DOR为 9.7 个月,中位 PFS 为 8.3 个月,中位 OS 为 16.3 个月;
队列 1 中 14 例患者此前接受过曲妥珠单抗治疗,其 ORR 为 50%,中位 DOR 为 7.0 个月,中位 PFS 为 5.5 个月,中位 OS 为 14.9 个月。
在队列 2 中, ORR 为 14%,中位 DOR 为 6.2 个月,中位 OS 为 9.6 个月。
安全性方面,45 例入组患者全部纳入了安全性分析,在 37 例患者中观察到治疗相关的不良事件(TRAE)。最常见的 TRAE(任何级别)为天冬氨酸氨基转移酶升高、丙氨酸氨基转移酶升高、皮疹、贫血及输液相关反应。4 例患者报告了 5 次 3 级 TRAE,包括输液相关反应、肾鞘膜积液、输尿管狭窄、血压升高和肝功能异常。未发生 4 级或 5 级 TRAE。
截图来源:Insight 数据库
封面来源:企业 Logo
免责声明:本文仅作信息分享,不代表 Insight 立场和观点,也不作治疗方案推荐和介绍。如有需求,请咨询和联系正规医疗机构。
编辑:ccai
PR 稿对接:微信 insightxb
投稿:微信 insightxb;邮箱 insight@dxy.cn
优先审批ASCO会议临床结果抗体药物偶联物临床研究
100 项与 上海津曼特生物科技有限公司 相关的药物交易
登录后查看更多信息
100 项与 上海津曼特生物科技有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年08月29日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
药物发现
3
1
临床申请批准
临床1期
2
4
临床2期
临床3期
6
1
批准上市
其他
2
登录后查看更多信息
当前项目
| 药物(靶点) | 适应症 | 全球最高研发状态 |
|---|---|---|
Becotatug ( EGFR ) | EGFR突变的非小细胞肺癌 更多 | 临床3期 |
纳鲁索拜单抗 ( RANKL ) | 骨转移癌 更多 | 临床3期 |
谷美替尼 ( c-Met ) | 非小细胞肺癌 更多 | 临床3期 |
盐酸伊立替康脂质体(石药集团欧意药业) ( Top I ) | 转移性结直肠癌 更多 | 临床3期 |
多西他赛(白蛋白结合型) ( Tubulin ) | 鳞状非小细胞肺癌 更多 | 临床2/3期 |
登录后查看更多信息
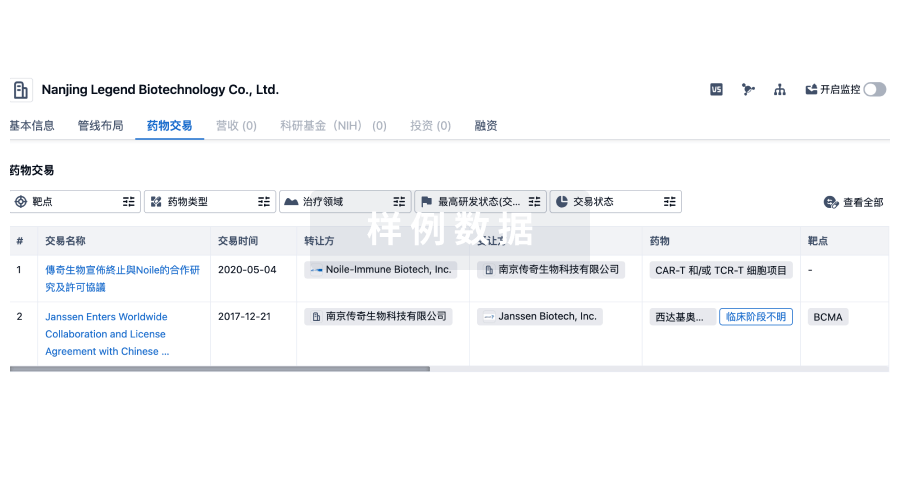
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
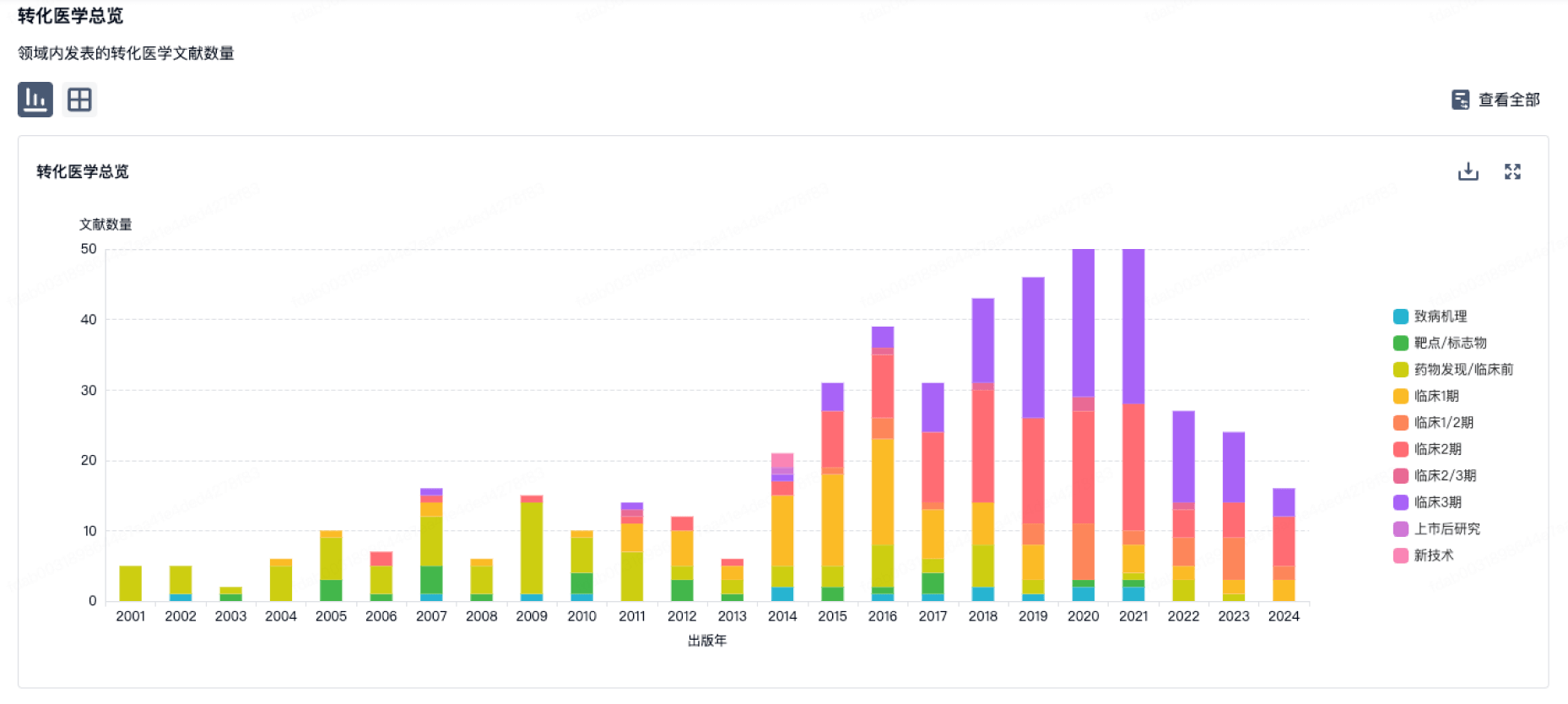
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
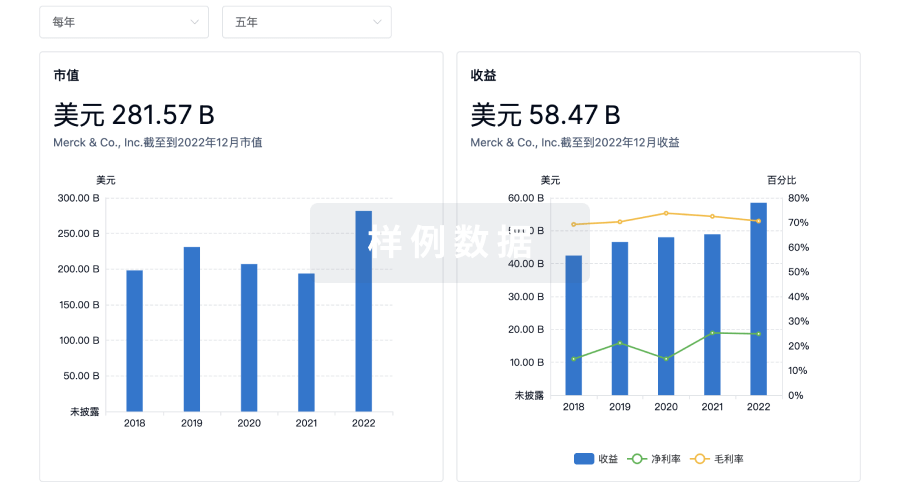
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
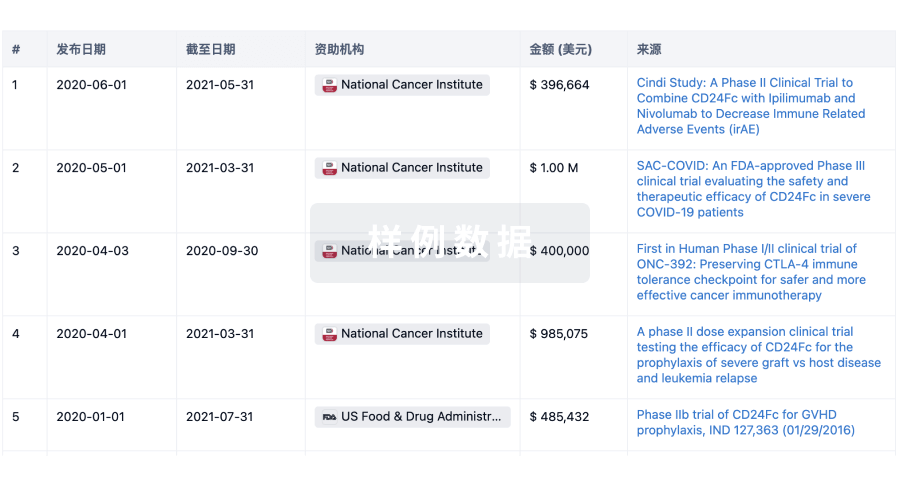
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用