预约演示
更新于:2025-05-07
Xinqiao Hospital
更新于:2025-05-07
概览
标签
其他疾病
感染
肿瘤
单克隆抗体
预防性疫苗
自体CAR-T
疾病领域得分
一眼洞穿机构专注的疾病领域

技术平台
公司药物应用最多的技术
靶点
公司最常开发的靶点
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
NCT06947694
A Prospective, Multicenter, Umbrella Design Clinical Study on the Abscopal Effect Induced by Different Radioimmunotherapy Combination Patterns in the Treatment of Non-Small Cell Lung Cancer With Multiple Metastases
NCT06953843
An Umbrella Trial of Combining Different Radiotherapy Fractionation Patterns With Immunotherapy for Multiple Metastases of Non-Small Cell Lung Cancer
NCT06943729
Clinical Safety and Efficacy of Flow Diverter in the Treatment of Intracranial Aneurysms
100 项与 新桥医院 相关的临床结果
登录后查看更多信息
登录后查看更多信息
2025-07-01Cancer Letters
Comparable survival outcomes in HLA-Matched and haploidentical hematopoietic stem cell transplantation for severe aplastic anemia patients aged 40–50: A CBMTR Registry-based propensity score matching analysis over the last decade
Article
作者: Li, Xin ; Zhang, Jian-Ping ; Zhang, Yu-Ping ; Xu, Ya-Jing ; Wu, Tong ; Wang, Chun ; Xu, Lan-Ping ; Zhang, Xi ; He, Peng-Cheng ; Zhang, Xue-Jun ; Ran, Xue-Hong ; Liu, Dai-Hong ; Huang, Jin-Xiong ; Xu, Zheng-Li ; Wang, Shun-Qing ; Su, Nan ; Lu, Pei-Hua ; Liu, Li ; Bai, Guan-Chen ; Zhou, Fang ; Chen, Jie-Ping ; Zhao, Xin ; Ye, Bao-Dong ; Yang, Hai-Ping ; Sun, Zi-Min ; Guo, Zi-Wen ; Su, Guo-Hong ; Zhou, Ming ; Yi, Hai ; Fan, Sheng-Jin ; Zhang, Yan-Ming ; Jiang, Er-Lie ; Song, Xian-Min ; Huang, Xiao-Jun
2025-07-01European Journal of Medicinal Chemistry
Design, synthesis, and biological activity study of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives against multidrug resistance in Eca109/VCR cells
Article
作者: Gong, Liang ; Guan, Yu ; Ouyang, Qin ; Li, Yu-Long ; Yu, Tao ; Lai, Wen-Jing ; Liu, Hong-Yuan ; Liu, He ; Xu, Bo ; Zeng, Rong
2025-05-01Bioactive Materials
A natural biological adhesive from slug mucus for wound repair
Article
作者: Jiang, Shichao ; Song, Jiahui ; Han, Lu ; Wu, Siyuan ; Fu, Liwen ; Mo, Xiumei ; Abdulhameed, Meera Moydeen ; Feng, Hao ; Shafiq, Muhammad ; Yuan, Zhengchao ; El-Newehy, Mohamed ; Xu, Yuan ; Wang, Zewen ; Wang, Xinyi
100 项与 新桥医院 相关的药物交易
登录后查看更多信息
100 项与 新桥医院 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年11月11日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
临床前
2
1
临床2期
其他
2
登录后查看更多信息
当前项目
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
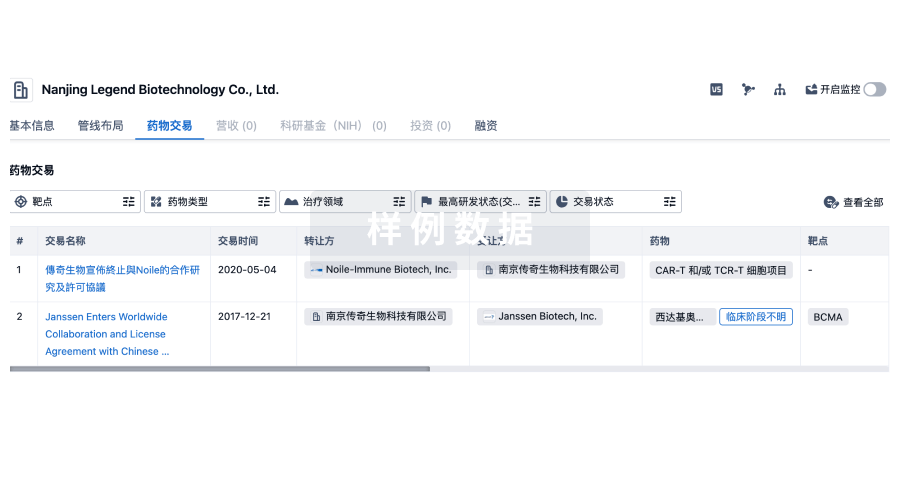
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
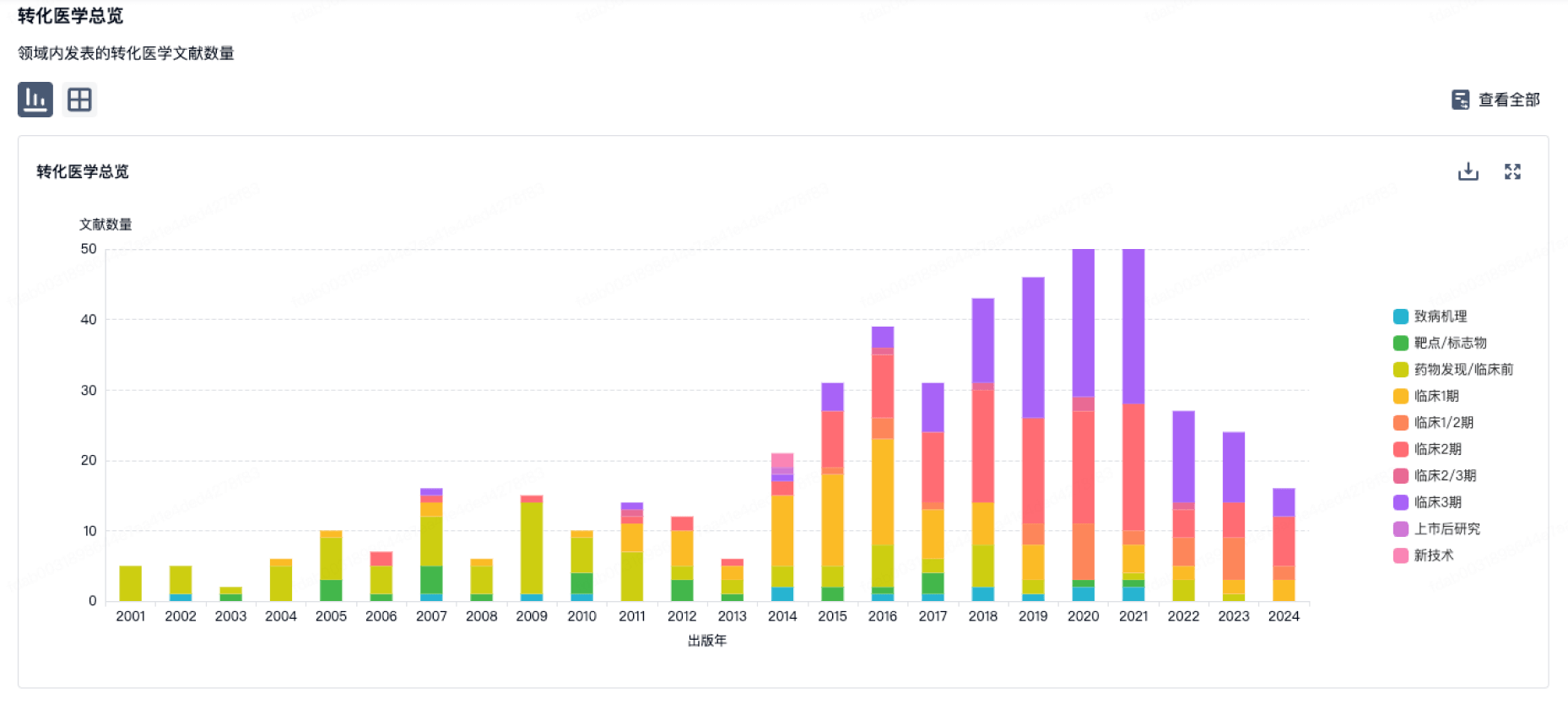
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
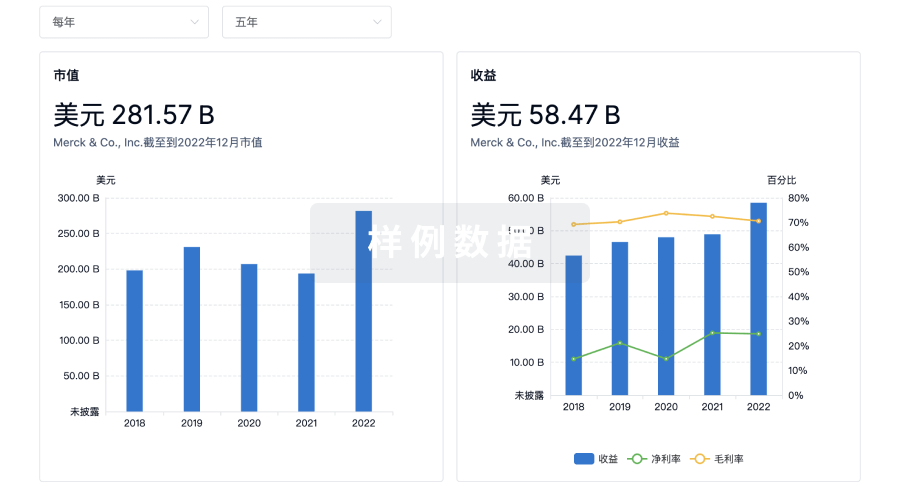
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用