预约演示
更新于:2025-08-29
Nuance Pharma (Shanghai) Co., Ltd.
更新于:2025-08-29
概览
标签
呼吸系统疾病
小分子化药
疾病领域得分
一眼洞穿机构专注的疾病领域

暂无数据
技术平台
公司药物应用最多的技术
暂无数据
靶点
公司最常开发的靶点
暂无数据
| 疾病领域 | 数量 |
|---|---|
| 免疫系统疾病 | 1 |
| 排名前五的药物类型 | 数量 |
|---|---|
| 小分子化药 | 2 |
| 排名前五的靶点 | 数量 |
|---|---|
| PDE3 x PDE4 | 1 |
关联
2
项与 优锐医药科技(上海)有限公司 相关的药物作用机制 PDE3抑制剂 [+1] |
在研机构 |
最高研发阶段批准上市 |
首次获批国家/地区 美国 |
首次获批日期2024-06-26 |


靶点- |
作用机制- |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症- |
最高研发阶段批准上市 |
首次获批国家/地区 中国 |
首次获批日期2005-10-28 |
4
项与 优锐医药科技(上海)有限公司 相关的临床试验NCT05743075
A Phase III, Randomized, Double-Blind, Placebo-Controlled Clinical Study to Evaluate the Efficacy and Safety of Ensifentrine Over 24 Weeks in Patients With Moderate to Severe Chronic Obstructive Pulmonary Disease
This is a multi-center, randomized, double-blind, placebo-controlled, parallel-group clinical study, which aims to evaluate the efficacy, safety, and PK characteristics of Ensifentrine 3 mg twice daily (BID) for 24 weeks treatment of moderate to severe COPD.
开始日期2023-03-10 |
申办/合作机构 [+1] |
NCT05758428
A Phase 1, Open-Label, Single and Multiple Dose Study to Evaluate the Pharmacokinetics, Safety and Tolerability of Nebulized Ensifentrine in Healthy Chinese Subjects
This is a phase 1, open-label, parallel cohort study to evaluate the PK, safety and tolerability of nebulized ensifentrine following administration of single and multiple doses in healthy Chinese male subjects.
开始日期2023-03-06 |
申办/合作机构 |
NCT05382546
A Phase 1, Open-label Pharmacokinetic Study of Intravenous NTM-001 (A Novel Formulation of Ketorolac Tromethamine Applied by Continuous Intravenous Infusion From A Pre-Mixed Bag) in Healthy Chinese Subjects
This will be a single center, open-label phase 1 study in healthy Chinese subjects. A total of up to 16 subjects may be enrolled to achieve a total target of 12 evaluable subjects. A full dose for NTM-001 will be defined as a 12.5 mg IV Loading Dose + 24-hour IV infusion (3.5 mg/hour).
开始日期2022-05-23 |
申办/合作机构 |
100 项与 优锐医药科技(上海)有限公司 相关的临床结果
登录后查看更多信息
0 项与 优锐医药科技(上海)有限公司 相关的专利(医药)
登录后查看更多信息
160
项与 优锐医药科技(上海)有限公司 相关的新闻(医药)2025-08-19
重磅路演推荐
项目名称:靶向恶性肿瘤干细胞的创新抗癌药
项目所在地:上海
适应症:脑胶质瘤、胰腺癌等恶性肿瘤
项目需求:
为两个新药管线的临床一期试验募集4000万资金。
公司简介
上海施江生物科技有限公司基于恶性肿瘤干细胞的全新特异性靶点,研发可安全有效杀伤肿瘤干细胞的线粒体特异性抑制剂,以解决恶性肿瘤复发这一临床难题,有望显著延长患者生存期。在研药物可用于脑胶质瘤、胰腺癌、三阴乳腺癌、小细胞肺癌等多种恶性肿瘤,具有广谱抗癌属性。公司计划于2025年下半年提交脑胶质瘤药物的中美IND申请,进入临床研究阶段。施江生物致力于为尚无有效治疗手段的恶性肿瘤提供创新解决方案,成为新一代肿瘤代谢类药物研发的引领者。
项目名称:安全长效的增肌减脂双适应症ASO
项目所在地:深圳
适应症:增肌、减肥、代谢性脂肪肝MAFLD、心力衰竭
项目需求:
1、需求:2000-2200万
2、投资架构:比较开放,可以是newCo,lisence out,共研co-dev,或者是平台公司加轮。
3、融资目标:全力推进项目至IND阶段,达成临床转化的关键里程碑。
公司简介
深圳市硬核酸生物科技有限公司,是一家创新核酸药物开发企业。基于物理第一性原理与全球领先的AI模型,公司优化反义寡核苷酸ASO的ADME特性,实现无需LNP包被或配体介导的器官靶向富集。目前,平台已在肌肉、脂肪、肾脏、肝脏、肺及中枢神经系统等器官取得显著成果。公司自创立以来,持续获得全球顶尖IT/BT企业与药企的战略关注及孵化支持。核心业务聚焦慢性病治疗领域,同时提供混合型服务。
项目名称:吸入双抗和三抗的创新之路
项目所在地:深圳
适应症:哮喘和慢阻肺
项目需求:
融资,合作开发和对外授权
公司简介
瑞思普利是一家集研发、生产、销售为一体的综合性创新型生物制药企业。公司目标是成为吸入药物研发和生产的领军企业。自研产品包括高端复杂吸入粉雾剂管线,重点领域聚焦在呼吸系统疾病,如哮喘,慢阻肺,特异性肺纤维化和肺动脉高压的吸入粉雾剂药物的仿制和创新药的研发。此外,公司还可以对外赋能:接受高端复合吸入粉剂(OEM)代工制造的委托;吸入型产品的定制开发(将常规剂型转化为可吸入剂型); 新型分子实体吸入药物递送系统的概念验证研究。
河络新图生物科技(上海)有限公司
项目名称:iPSC分化的异体血小板的研发
项目所在地:上海
适应症:化疗引用的血小板减少、外科手术、原发性血小板减少
项目需求:
1.共同开发iPSC衍生的血小板
2.创新止血和抗血小板新药的早研
3.人造血小板与其它止血剂的联合应用
4. 人造血小板及衍生产品对外授权
5. 人造血小板衍生产品在医美领域的合作与开发
公司简介
河络新图成立于2021年,创业团队由美国麻省理工学院和英国剑桥大学归国学者领衔,是一家定位于iPSC定向分化、基因编辑、规模化细胞产品制备及细胞治疗技术开发为目标的高科技创新型企业。河络新图在iPSC领域以多年创新的细胞定向分化技术解决再生医学领域的核心问题和临床需求, 倾力打造人工血液制备平台, 同步建设全球领先的再生医学细胞产品开发体系。公司研发团队由来自于海内外著名高校和研究机构的青年学者和拥有多年产业经验的杰出人才组成,团队在干细胞、转化医学领域均拥有丰富的科研及产业经验。公司正在开展人工血小板的研究者发起性临床试验,初步结果显示产品安全有效。
浙江霍德生物工程有限公司
项目名称:针对脑卒中、颅脑损伤等运动障碍后遗症的异体通用型iPSC衍生神经前体细胞治疗产品
项目所在地:中国杭州市,美国休斯敦
适应症:缺血性脑卒中偏瘫后遗症、出血性脑卒中偏瘫后遗症、颅脑损伤后遗症、脑瘫、癫痫
项目需求:
1. 商业合作:寻求与全球知名药企的hNPC01产品的海外共同开发或商业化授权合作,加速产品上市及销售。
2. 融资:近期将完成C轮融资,用于支持hNPC01产品的海外多中心的确证临床和商业化。
3. 宣传:对于脑卒中等未被满足的巨大临床需求和潜在重磅细胞药物临床突破性数据、加速路径等进行宣传;欢迎更多优秀的人才加入和各种合作。
公司简介
霍德生物于2017年1月成立,是一家专注于开发iPSC衍生通用型神经细胞治疗产品的全球创新生物技术公司,在杭州拥有细胞药物研发中心、GMP细胞生产车间与质控中心和办公场地。霍德生物在iPSC重编程、多能干细胞神经分化及细胞工程方面具有领先的全球专利及创新技术优势,建立了iPSC细胞产品CMC平台,悬浮自动生产工艺和多种创新分析方法。霍德生物拥有多款神经细胞在研产品,其中首个人前脑神经前体细胞注射液hNPC01是全球首个获中美注册临床默许的多能干细胞品种,已完成针对脑卒中偏瘫后遗症的中国I期注册临床,治疗效果支持其为潜在的BIC,可拓展颅脑损伤后遗症、脑瘫、癫痫等重大未被满足的临床需求。霍德生物成立以来已完成多轮由高瓴投资、礼来亚洲等领投的数亿元融资。
河南知微生物医药有限公司
项目名称:1类新药BTK抑制剂TM471-1的研究与开发
项目所在地:河南省新乡市凤泉区产业集聚区
适应症:非霍杰金淋巴瘤及自身免疫性疾病
项目需求:
1.融资需求:计划融资 15000 万元,主要用于BTK抑制剂TM471-1后续临床试验推进、研发团队扩充以及生产设施筹备等方面,加速产品上市进程。
2.合作拓展:与上海及长三角地区的科研机构、高校、药企等建立合作关系,实现资源共享、优势互补,共同开展新药研发、临床试验合作等项目。
3.对接BD:与相关国内外BD人士对接,寻求项目License out。
公司简介
河南知微生物医药有限公司于 2016 年 11 月 15 日成立,总部位于新乡市凤泉区产业集聚区,在上海张江药谷建有研发中心。
公司由新乡王氏集团与李伟教授和留美博士丁清杰(原 Roche 首席科学家)科研团队联合创建,并与河南师范大学达成战略合作。其业务主要涵盖抗癌类创新药物研发、高端原料药及其制剂研发,以及相关领域技术服务。在科研实力上,组建了包含博士、硕士等50多人的专业团队,打造了1类创新药从药物发现、工艺优化、临床前研究、临床试验、申报注册等各个环节全贯通的高端创新技术平台,配备先进实验仪器,建成超 6000 平方米综合实验室。公司已取得5项 PCT 国际专利,拥有三个一类创新药临床批文。先后获得河南省新型研发机构、河南省抗癌新药工程研究中心等资质。
项目名称:Pan-RAS (ON) Tri-complex Molecular Glue Inhibitor
项目所在地:上海
适应症:实体瘤
项目需求:本项目聚焦 pan-RAS 通路创新药物开发,目前处于临床前研究阶段,已完成靶点验证、化合物筛选及初步药效学研究。项目核心优势包括:针对 RAS 突变(如 G12C/D/V、G13D等热点亚型)的广谱抑制机制,通过高选择性小分子抑制剂突破传统 RAS 靶点 “不可成药” 瓶颈;在多种肿瘤模型(如 NSCLC、PDAC、CRC)中展现显著抗肿瘤活性,联合用药方案(如与免疫检查点抑制剂联用)可进一步增强疗效;药代动力学特性优异,具备口服生物利用度高、体内代谢稳定等成药潜力。
现寻求全球范围内的合作伙伴,合作模式包括但不限于:海外市场权益转让、全球联合开发、技术共建及商业化合作。期望合作伙伴具备以下条件:在肿瘤领域有丰富的药物开发经验及国际化注册能力,拥有成熟的临床试验网络及商业化渠道,可提供资金、技术或资源支持以推进项目临床转化。项目方将提供完整的临床前研究数据(含药效、药代、安全性评价报告)及技术转移支持,与合作伙伴共同探索 pan-RAS 靶向药物的全球市场价值,推动创新疗法惠及更多肿瘤患者。
公司简介
荣昌制药成立于1993年,总部位于中国烟台,是一家集研发、生产、销售、服务为一体,横跨现代中药、小分子和生物制药、生物药物研发和生产服务(CDMO)、生物医药企业和项目孵化四大领域的现代化制药企业。公司的创始人和管理团队拥有多年顶尖跨国药企的研发和管理经验,并参与了多个临床及上市新药的研发。多年来成立的子公司和分拆公司包括荣昌生物、业达国际生物医药创新孵化中心、烟台迈百瑞国际生物、烟台赛普生物、荣昌制药(淄博)等。荣昌制药小分子研发板块专注于肿瘤新药研发,以肿瘤靶向精准治疗和肿瘤免疫治疗药物为核心,致力于开发生物学机制清晰,具有重大未满足临床需求的潜在first-in-class或best-in-class创新药物,现已形成涵盖实体瘤、血液瘤的研发管线布局。
项目名称:转录因子蛋白质原创药诱导细胞转化癌症治疗项目
项目所在地:江苏省苏州市
适应症:肝癌、胃癌
项目需求:苏州复恩特药业(美国Qurgen中国全资子公司)依托全球首创“QQ-蛋白质诱导原位组织重编程技术”,开发不杀死癌细胞而将其转化为正常细胞的颠覆性疗法。现寻求战略投资方、跨国药企BD合作及临床资源支持,加速中国临床试验进展及产业化进程。
1、核心需求如下:
1) 第二轮融资1.5-2亿人民币
估值基准:投前15亿人民币。
用途:
✔ 80%用于中国一期临床(肝癌/胃癌适应症)试验,进入二期临床试验;
✔ 20%投入储备项目的研发(糖尿病、慢性心衰、卵巢早衰、肺纤维化等项目);
2) 寻找战略合作开发伙伴:开展深度合作,采用:授权开发、共同开发的形式,共同开发新药管线。
2、技术优势背书
全球唯一转录因子成药的临床转化:SON-DP注射液获FDA/CDE双批准,实现“肿瘤组织原位转化”(活检显示35%-100%癌组织转正常);
专利壁垒:3项核心技术专利(零竞品对标)。
公司简介
苏州复恩特药业是一家专注于蛋白质生物制药的企业,着重于开发原创转录因子抗肿瘤药物和转录因子药物治疗目前无根治疗法的重大疾病。 公司的创新型转录因子抗肝癌、抗胃癌蛋白质药物是一款诱导体内肿瘤组织原位重编程药物。其原理是将癌细胞转化为正常细胞,从而治疗实体瘤,为癌症患者提供安全、有效且可负担的治疗方案。2024年10月15日,公司已获得中国国家食药局药审中心一期临床试验许可(IND)。2025年3月,公司已启动抗肝癌药、抗胃癌药一期临床试验,目前已开展6例患者给药。凭借具有自主知识产权的突破性技术平台,苏州复恩特药业正在领导开创抗肿瘤药物临床研究和晚期肿瘤治疗的新时代。
更多路演申请
路演时间:2025年8月29日
路演形式:路演-需求快闪
路演-早期/晚期项目
每位项目方15mins
参会人群:公司管理层:董事长、总裁、首席执行官、总经理、首席战略官、首席商务官、首席投资官、首席医学官、院长、协会会长、合伙人
申报截止时间:2024年8月22日
报名抢占黄金席位即可获得
跨国药企战略合作直通车
让您的创新项目一键接入全球TOP药企资源库,开启跨国技术合作与联合研发新机遇。
顶级投资机构优先观察名单
直通头部资本视野,加速融资进程,抢占生物医药赛道制高点。
全球交易范式全解析
深度掌握前沿BD模式、跨境并购策略与IP授权逻辑,解锁生物医药产业全球化布局密码。
2025(第九届)亚太生物医药合作峰会
全球协同创新 多元开放合作
2025年8月28日-29日 中国 · 上海
生物医药 BD 领域风云际会,波澜壮阔。在这种中国资产迈向国际化的大时代下在这样一个狂风骤雨的时代环境中,药企如何加速国际化进程?如何通过多元化合作,加速协同创新?盈利曙光下,创新药企如何通过现金流大考?--都成为企业不得不思考的问题。
基于此,由上海士研咨询主办,苏比亚SUBIA和雅法资本支持举办的Bio PartneringAPAC 2025(第九届)亚太生物医药合作峰会以“全球协同创新,多元开放合作”为主题,定于2025年8月28-29 日·上海(周四、周五)召开。重点聚焦药企如何在全球化浪潮中把握确定性,实现国际化战略宏图,以寻求新的增量;加快药企国际市场布局,深度探索产品商业化的机遇与挑战;同时,我们也会邀请来自亚洲各国家乃至全球的最前沿技术的创新药企参与,分享各自国家和地区的先进技术和成功案例,积极寻求更多全球合作新机遇,深度探索“合作”为医药生态圈带来的更多可能。
扫描上方二维码咨询报名/赞助/合作/路演
工作人员会在1-2天内联系您
已确认嘉宾信息
刘东舟
首席科学官、创新药全球研发中心总经理,华东医药股份有限公司
南开大学化学/生物化学/免疫学学士、博士,美国纽约州立大学石溪分校 MBA,曾在美国北卡大学教堂山分校药学院、美国肯塔基州路易斯维尔大学医学院等进行医学、药学博士后研究。具有30年海内外创新药研发工作经验,在药物发现、转化医学和临床开发积累了丰富的经验。曾在美国GSK(葛兰素)、Wyeth(惠氏)等公司担任关键研发部门负责人。2020年加入华东医药担任公司首席科学官兼创新药全球研发中心总经理,负责创新药研发工作。
曾发表130多篇研究文章/学术会议摘要及主题演讲,申请50余项创新药专利(8项授权)。海外高层次人才认定专家;浙江省“领军型创新团队”带头人,主持多项政府科技项目;中国药促会专委,中科院合肥物质所特聘教授,浙大特聘客座教授、(国家级)博士后科研工作站导师。
陈波
首席科学家、科委会副主任,华润医药集团
陈波博士是海外高层次人才特聘专家,目前担任华润医药集团首席科学家, 在生物技术和制药行业拥有超过20年的国际职业生涯,包括在美国NIH,AstraZeneca等顶尖科研机构和制药公司从事基础研究和新药研发团队领导者的工作经历,具有从靶点发现到临床开发和商业运作的整个研发价值链的研发背景。他博士毕业于中国人民解放军空军军医大学,在美国NIH完成博士后,其后还获得美国Hood College工商管理硕士学位。他迄今在著名期刊上发表国际学术论文,专著章节和专利共30余篇,包括发表在Annals of the Rheumatic Diseases, Nature Communication, Nature Immunology, Nature Medicine, PNAS 等顶尖杂志上的论文。并曾获得了卫生部的科技进步二等奖,AstraZeneca的员工白金奖和银奖,NIH的卓越研究员奖,齐鲁制药集团的卓越领导奖等。
周高波
首席投资官,先声药业
周高波,先声药业首席投资官,负责公司对外合作和战略。加入先声药业之前,周高波先生在麦肯锡公司担任全球董事合伙人和大中华区医疗咨询联席负责人,拥有十五年医疗行业管理咨询经验。他一直致力于为领先的制药、生物科技、医疗器械、生命科学投资公司提供战略和管理咨询服务,深入探索中国医改和创新环境下的企业战略、创新商业模式、数字化转型、投资合作,并组建了行业内最大的医疗领域管理咨询团队。此前,他在人类基因组科学公司(HGSI)亦从事过抗体和融合蛋白药物的研发工作。
周高波先生拥有杜克大学商学院的工商管理硕士学位,马里兰大学生物化学硕士学位和复旦大学的遗传学本科学位。
佟玲
BD负责人/ 创新拓展副总经理,步长制药
理学博士,国家“131”第一层次创新人才,拥有16年医药研发经验,10年BD交易和联盟管理经验。曾在天士力医药主导完成与法国Pharnext、美国礼来、日本EA等国际合作。加入步长后,组建了生物药BD团队,推动BC001、Fc-EPO等项目成功出海到俄罗斯、东南亚等国家,并主导了步长与天坛神经外科研究所、佑嘉生物、美国REMD等机构的战略合作。在BD规划、产品线优化以及联盟建设领域积累了丰富经验。
许倩
医学副总裁,金赛药业
许倩博士毕业于新加坡国立大学,取得医学博士学位,并在加州大学完成博士后培训,拥有10多年免疫、皮科、心血管相关治疗领域的临床开发经验。
许倩博士在金赛药业负责免疫,皮科,眼科,成人内分泌,女性健康等治疗领域的临床开发工作。在此之前,许倩博士曾就职于罗氏、礼来、诺华、恒瑞医药等大型国际制药公司参与新药临床开发工作。经验涵盖产品的早期评估、CDP 制定、从 FIH到 NDA及上市后医学的全周期。 在加入工业界前,许倩博士有多年临床和科研经验。
曹飞
联合创始人兼首席运营官,应世生物科技(南京)有限公司
曹飞,应世生物联合创始人兼首席运营官,中国生物工程学会精准医学专委会副秘书长。他在医药领域拥有超过近20年的运营、商务拓展与管理工作经验,2015至2018年,他创立并担任北京鼎基生物科技有限公司首席执行官。在此之前,他于2011加入百济神州(北京)生物科技有限公司并担任公司高层管理委员会成员。他的履历还包括博奥生物集团投融资部副总监、葛兰素史克公司大中华区业务拓展经理以及杨森制药公司亚太区业务拓展与外部科学事务负责人等职务。
他本科毕业于北京大学,获生命科学学院和中国经济研究中心双学位。他曾在香港科技大学生物系攻读博士学位,并赴美国宾夕法尼亚大学医学院做访问学者。
杨笛
董事、副总裁,翰宇药业
杨笛,生物科学学士、生物医学硕士、金融财务硕士。拥有超过10年生物制药和医疗器械领域研发、风险投资、证券、及BD经验。2017年加入翰宇药业,历任战略投资高级总监,现任本公司董事、副总裁、董事会秘书,分管证券、BD。
邱婧君
合伙人、全球研发中心副总裁、 生物统计与数据科学部总经理,复星医药
邱婧君博士曾就职于美国耶鲁大学医学院、默沙东(美国Merck)、拜耳和百济神州。
具有不同地区、不同治疗领域临床研究的丰富经验;参与从研发战略规划到临床试验及上市后研究的各重要阶段,包括早研转化与BD尽调项目。十几年来一直组织推动研发试验设计与统计社区的各种活动。
现任中国统计理论与方法专委会、药促会、CSCO等统计专委会委员,并支持不同课题工作小组,包括ICH-E9(R1) WG、Global IFPMA ICH-E20 WG;与多所科研机构建立长期交流合作关系。
姜大为
业务发展总监,上海医药集团业务发展部
上海医药集团业务发展部工作期间,协同各业务团队领导同事,广泛接触各适应症领域和开发阶段的创新药项目和公司,参与完成多项创新和成熟药品项目的对内&对外授权合作项目。加入上海医药集团之前,曾在多家国内生物医药上市公司从事创新药跨境BD和相关工作,参与完成了多个创新药项目BD交易。
高建英
副总裁兼战略联盟管理部及卓越上市部总经理,复星医药
高建英(Jenny Gao)女士现任复星医药战略联盟部总经理及新产品卓越上市部总经理。
高建英(Jenny Gao)女士具有近20年医药行业工作经验,在心血管慢病及特药肿瘤领域负责个多个重磅产品上市及卓越营销,并擅于国际战略合作及产品全生命周期管理。
黄薇
肿瘤领域首席医学官,石药集团
拥有25年临床经验,包括15年全球药物研发行业经验和10年临床实践经验。
资深肿瘤科医生,专注于全身系统治疗和放疗,具有10年以上临床实践经验
15年以上行业临床开发经验。先后供职于MerckSerono 1.5年,Roche 7年,BeiGene 3年,Simnovabio 1年。具有临床开发职能团队的建立和建设、人员管理、全球产品开发策略、全球申报(FDA/EMA)、中国申报、试验设计、临床试验医学监查和与研究专家有效互动的经验。在10个以上的肿瘤项目(从Ph I到III)和上市后做出过杰出贡献。精通监管指南,具有与卫生当局互动的丰富经验,并与美国、欧盟和亚洲的KOL合作和建立了良好的联系。
许笛
拜耳Co.Lab中国负责人,拜耳处方药事业部
许笛博士拥有十年风险投资经验及数年肿瘤,CGT领域研发经验,目前担任拜耳Co.Lab中国负责人。作为拜耳面向全球生命科学领域的共创平台,拜耳Co.Lab旨在通过促进生物技术生态的开放合作,已完成在包括中国、美国、德国、日本等全球创新热点地区的战略布局。拜耳Co.Lab在中国计划赋能10至15家本地初创企业,重点聚焦肿瘤、心血管、新技术平台以及细胞与基因疗法等领域的前沿创新,通过提供理想的共创空间和专家辅导赋能初创企业,将突破性创新成果转化为具有深远影响的医疗健康解决方案。
马大海
VP、大中华区业务拓展负责人,和铂医药
神经外科医师,在医药领域有20余年的经验,先后就职于诺华制药,拜耳医药保健有限公司,默克医药担任不同BD管理职位,曾任上市公司天士力医药集团BD负责人,目前在和铂医药担任大中华区BD负责人。有从早期直至商业化阶段全生命周期产品的BD经验,具有少数股权投资和合资合作,New Co等实操经历。
拥有北京大学医学部临床医学硕士学位,欧洲工商技术管理学院(ESMT)MBA学位。
李果
中国商业数字化转型负责人,葛兰素史克(中国)投资有限公司
拥有超过20年的企业数字化转型、全渠道营销和信息技术管理经验。先后在汽车、快消、医药领域的世界五百强工作,兼具本土和海外经历。在数字化研发和营销、精益创新、孵化新业务以及业务模式转型等方面有丰富的实践积累。
唐凡
数据卓越部门负责人,勃林格殷格翰
唐凡博士在美国Univercity of Iowa 获得生物统计博士学位。毕业后首先加入基因泰克美国作为临床统计师参与不同治疗领域的研发工作。自2019年7月入职勃林格殷格翰中国,带领亚太区健康信息科学团队,探索真实世界数据以及证据在临床研发全生命周期的应用。自2021年1月起,带领中国数据卓越团队,协助公司搭建行业领先的基于云的大数据管理分析平台,协助公司转变成洞察驱动的组织。在2023年10月开始,同时监管数据卓越团队,数字化团队,以及总部上市推广赋能团队,支持公司以数据驱动,围绕数字化的技术,赋能业务部门在整个商业生命周期的转型和变革,实现最佳的客户体验。
魏志军
全球药品开发分析部统计编程与创新副总监,诺华制药
Stanley(魏志军)毕业于复旦大学药学院,药理学硕士学位,诺华制药统计编程副总监,目前专注于创新与工具开发,18年行业工作经验,熟悉临床试验全流程,擅长工具、软件和应用的开发,并对开源软件技术有着深入的研究与应用。Stanley 对人工智能(AI)在临床试验中的应用充满热情,并积极探索其潜力。他曾在临床试验数据标准协调委员会(CDISC)中国区交流大会分享了《关于AI智能体在临床数据运营中的创新应用》,展示了如何利用AI技术优化数据管理和分析流程。此外,他还作为联席主席主持了PhUSE 2024 上海 SDE(Single Day Event),围绕主题《解锁人工智能在临床试验中实现端到端革命性处理》,Stanley也是今年8月制药行业中国区编程大会(PharmaSUG)AI会前培训负责人,助力推动AI技术在药物研发,尤其是临床试验中的前沿实践与行业交流。Stanley作为CDISC 中国 C3C核心成员,也曾作为特邀外部专家, 共同参与国家CDE《药物临床试验数据递交指导原则》的制定工作。
沈蓉
中国业务发展转型负责人,辉瑞投资有限公司
沈蓉女士在医疗产业、投资及咨询领域拥有多年工作经验,现任辉瑞中国业务发展转型负责人。在加入辉瑞之前,她曾在晨兴创投等知名投资机构任职,从事创新药和新兴技术的投资,并于IQVIA等咨询公司任管理咨询顾问,为跨国药企和本土生物科技公司提供战略支持。沈蓉女士于中国科学技术大学获得化学专业学士学位,后于宾夕法尼亚大学获得生物化学博士学位。
肖申
首席科学官,礼邦医药
肖申博士现任礼邦医药首席科学官负责公司的非临床,临床开发和全球药物注册工作。之前任海森生物首席医学官,思路迪医药首席医学官和首席战略官。肖申博士曾在美国食品药物监督管理局(FDA)担任临床高级审评员负责心、肾、血管疾病新药的临床审评。在近二十年的 FDA工作生涯中,负责审评了数百个新药开发(IND)的各个阶段,跨度从临床前期、临床各期、及上市后的药物疗效、安全跟踪、随访;做为临床评审员和/或综合评审小组负责人负责审批了十几个新药的上市工作。其间曾在 FDA 医疗器械审评中心(CDRH),负责多个相关医疗设备的审评、审批(IDE,510k,PMA)。
加入 FDA 之前,曾作为内科临床主治医生、博士研究助理和博士后有十多年的临床工作和实验室研究经验,包括各类普通内科疾病诊治、抗生素临床药代动力学的研究、细胞信号传导的研究等。其间除了曾获国家科技进步三等奖一项,全军科技进步二等奖一项外,还获得 FDA,美国生理协会(APS),国际肾脏病协会(ISN),日本透析和人工脏器协会的多项奖励。
吴辰冰
创始人兼首席执行官,上海岸迈生物科技有限公司
吴辰冰博士从美国佐治亚大学获得生物化学与分子生物学博士后,进入美国哈佛医学院进行博士后研究,后来进入雅培制药公司生物研究中心,从事治疗性抗体的研究与开发十多年,带领多个项目从早期研究开始成功进入到临床申报,有多篇首席作者论文发表在国际一流学术刊物,并有多项国际专利授权。之后吴博士回国加入睿智化学和三生国健,完善研发梯队,建立技术平台,成功地开发出一些有影响力的创新抗体药物。2016年5月吴博士创立了岸迈生物,主要进行新一代生物大分子的技术开发及产品研发,带领团队开发出新一代的双特异性抗体平台技术“FIT-Ig”,目前平台技术专利已获多个国家专利局授权,已有由该技术开发出来的新型双抗项目进入临床I/II期,未来将致力于建立一个创新型国际化的生物制药企业。
刘利平
创始人,深圳君圣泰生物技术有限公司
刘利平博士是港股上市公司君圣泰医药(2511.HK)的创始人&首席执行官及董事会主席;君圣泰医药是一家全球一体化的新型生物技术公司,专注代谢性疾病等领域的重大未满足临床需求。以“标本兼治、深求治愈”这一DeepCure理念为目标,公司立足源头创新,开发First-in-Class的原创新药,以期为全球患者提供基于中国智慧的,安全、有效、综合获益的解决方案。基于自主知识产权,公司已构建丰富产品管线,在全球推进多项中、后期临床试验,开发代谢相关脂肪性肝炎(MASH)、2型糖尿病(T2DM)、严重高甘油三酯血症(SHTG)和原发性硬化性胆管炎(PSC)等适应症。作为同类首创的多靶点新分子实体,熊去氧胆小檗碱(HTD1801)被美国FDA授予2项“快速通道资格认定”、1项“孤儿药资格认定”,并获得国家“十三五”“重大新药创制”科技重大专项支持。
朱凌宇
首席商务战略与发展官,启德医药科技(苏州)有限公司
朱凌宇博士曾先后在跨国制药企业(美国辉瑞公司和强生公司)和本土创新制药企业(贝达药业和赛生药业)等多家知名药企负责 BD业务。拥有 28年的国际医药行业工作经验,主持完成了40项总价值超过178亿美金的战略交易,其中包括了为贝达药业引进了安进公司的上市肿瘤产品帕妥木单抗,和美国Xcovery公司的临床肿瘤项目恩沙替尼。 恩沙替尼已于2020成功上市,成为贝达药业继埃克替尼之后第二个打破跨国药企垄断的国产抗肿瘤创新药产品。也包括他为赛生药业在18个月连续引进了意大利美纳里尼公司两款在美国上市的创新药产品--Vaborem和艾拉司群。艾拉司群, 作为目前全球首个获批的口服SERD乳腺癌药物,有望成为赛生继日达仙之后的又一重磅产品。2024年,朱博士加入了启德医药,至今已经完成了4项ADC的出海交易,包括与美国 Biohaven、韩国 AimedBio达成重大签约,总金额超130亿美金,创生物药出海新纪录。在2009年回国之前,朱凌宇博士曾担任美国Esperion生物制药公司商务拓展资深经理,推动完成了2003年底该公司与美国辉瑞公司的13亿美元现金的战略并购交易。
张弢
全球统计与数据科学部人工智能与机器学习高级总监,百济神州
张弢,现任百济神州全球统计与数据科学部人工智能与机器学习高级总监。他毕业于北京大学数学科学学院,在人工智能创新领域有超过20年经验。他专注于大语言模型,多模态模型,生成式基础模型,大数据与云计算系统的前沿探索。在加入百济神州之前,张弢曾任职于阿里巴巴,主导及参与智能驾驶、车联网以及智能语音助手“天猫精灵”等开创性工作。除此之外,他曾担任生成式人工智能领域的资深顾问与导师,为医药、高端制造等科技行业提供专业咨询,助力其利用人工智能实现技术创新,提升运营效率。
卢安邦
执行董事兼首席执行官,维昇药业
卢安邦先生在医药行业拥有超过30年的工作经验,目前担任维昇药业执行董事兼首席执行官。卢安邦先生在大中华区和欧洲都有丰富的工作经验。他曾在武田工作7年,先后担任中国区与大中华区总裁。在此之前,他在施维雅台湾、中国大陆和法国工作16年。更早期的职业生涯是在台湾阿斯利康公司。
卢安邦先生主导了武田公司在中国的关键成长期。武田中国在他的领导下,销售收入增长了超过10倍。作为对自己的挑战,卢安邦先生选择从零开始创建维昇药业,并将其打造成一家成功的生物医药公司,致力成为内分泌相关治疗领域的专家。
张彤
联合创始人,首席商务官兼首席财务官,橙帆医药
张彤博士,橙帆医药联合创始人、首席商务官与首席财务官,负责国内国际合作,融资及投资者关系。张博士曾任上海吉凯基因有限公司首席商务官,康桥资本董事总经理,参与了云顶新耀的孵化,并主导了应世生物和优锐医药的投资。
他曾任药明康德全球BD负责人,主管战略合作/投资,并购/孵化,执行了和梅奥诊所的JV药明奥测的成立,以及药明生物和美国Arcus公司PD-1项目转让; 默沙东中国BD负责人,执行了默沙东/先声合资企业建立以及和重庆智飞的疫苗战略合作关系。张博士在美国曾任默沙东新兴市场BD执行总监,EKR公司BD负责人,瑞士信贷(纽约)医药证券分析师,以及美国ISO Healthcare Consulting和Defined Health咨询顾问。
张博士毕业于武汉大学生物系,在哥伦比亚大学取得生物学博士,曾在纽约Sloan-Kettering 癌症中心诺贝尔医学奖得主James Rothman实验室从事博士后研究。
乔萌
总监,辉瑞(中国)研究开发有限公司
乔萌目前任职于辉瑞(中国)研究开发有限公司,是临床药理,临床生物分析/生物标记物 团队负责人,所带领团队支持公司全部研发管线产品临床I,II,III期及上市后研究。在入职辉瑞前,曾在强生(中国)研发中心,和记黄埔医药(上海)医药有限公司,中美冠科从事临床前及转化医学研究。
柳丹
管理合伙人,Pivotal bioVenture partners
柳丹博士现任Pivotal bioVenture Partners管理合伙人,负责中国区生命科学投资业务。在此之前,他曾任鼎晖投资基金高级合伙人/管理合伙人/投委,负责医疗大健康领域多只基金的投资,投管包括:Zenas(ZBIO.O)、艾博兹(被并购)、药康生物(688046.SH)、成都先导(688222.SH)、I-Mab(IMAB.O)、迈新生物(被并购)、和铂医药-B(2142.HK)、复诺健生物、新格元生物、齐碳科技、劲方医药等数十个优秀项目。
在加入鼎晖投资前,柳博士曾在Bain & Company主要参与大健康领域的管理咨询业务。期间,他服务并负责过多个大型跨国药械企业的战略项目。柳丹博士累计在科研、商业、管理、咨询、投资等多领域拥有近20年生命科学行业相关经验。
柳丹博士同时担任多个政府、行业协会理事和高级专家,及多个省市级科技和人才项目的审评专家;多次获评行业权威榜单“40位40岁以下投资人”、“年度最佳医疗大健康领域投资人物TOP 10”等荣誉。
沈丽丽
董事总经理,晨兴创投
沈女士具有20年生物医药/医疗健康领域的工作经验,目前担任十余家生物医药公司的董事。沈女士于2010年加入晨兴创投,主要负责生物医药的投资管理。沈女士是复旦大学工商管理硕士,西安交通大学生物化学与分子生物学硕士。
毛 化
合伙人&董事总经理,弗若斯特沙利文咨询有限公司
毛化先生是弗若斯特沙利文公司合伙人兼董事总经理,专注于医疗健康领域,负责公司大中华区医疗咨询和资本市场业务,业务涵盖生物科技,药品,医疗器械,医疗服务等各个细分领域。毛化先生拥有丰富的企业境内外上市、收购兼并、企业市场咨询等业务经验。毛化先生曾经参与主导了多个影响力较大的港股和美股上市项目,并和国内外领先的投资公司保持紧密的业务合作关系。
黄翰漾
医药行业首席分析师,兴业证券
黄翰漾,兴业证券医药行业首席分析师。2016年加入兴业证券医药团队。医药行业全覆盖,曾负责过创新药产业链、医疗器械、综合类医药企业、中药等多个细分领域研究,同时负责医药产业政策研究并参与团队管理。兴业证券医药团队是市场上成立最早,规模及影响力最大的医药团队之一,截至2024年团队共计8次获评权威评选最佳分析师第一,累计上榜14次。
刘晓华
复健资本
新加坡国立大学博士,有20多年医药行业和基金从业经验,曾任上海医药工业研究院制剂研发科学家,BASF医药部门市场经理,复星医药国际部和BD部门投资总监,德邦证券星盛资本联席总经理。现任复健资本新药基金董事总经理。
肖汀
合伙人,康禧全球投资基金
肖汀自2015年加入康禧全球投资基金,是其创始团队成员和并担任合伙人。肖先生在生命科学投行和投资领域拥有17年的经验。
肖先生领导了公司对以下公司的投资并担任董事会成员:迪泰医学、诚益生物、立博医药、ReNiva Medical、STRM.Bio;肖先生还担任Allecra Tx、Atia Vision、Rgenta Tx和Sirius Tx的董事会观察员。肖先生此前还曾担任Zenas Biopharma(纳斯达克股票代码:ZBIO)、卢凯制药(被康哲药业收购)、三叶草生物(港交所股票代码:2197)的董事会成员,以及Apama Medical(被波士顿科学收购)的董事会观察员。
加入康禧投资之前,肖先生在中金公司和高盛集团投资银行部工作了7年。 肖先生拥有约翰·霍普金斯大学生物技术硕士学位和上海交通大学经济学学士学位。他还是特许金融分析师 (CFA) 持有者。
陈小五
化学副总裁,上海柯君医药
陈小五毕业于厦门大学化学系,1993获得美国宾州州立大学生物化学博士,随后在加州大学旧金山分校(UCSF)从事计算化学博士后研究,1997加入美国吉利德科学公司从事创新药研发,2018年任斯坦福大学医学院访问学者,目前在上海柯君医药公司从事创新药研发。 陈博士是创新药行业的老兵,有着~30年创新药研发经验,在吉利德19年期间,参与了多项抗病毒,肿瘤药物研发,包括世界第一个流感口服药达菲(奥司他韦)及多个“重磅炸弹级”的艾滋病和丙肝药物的研发。目前在柯君医药,参与了柯君医药新一代Best-in-Class抗血小板药物CG-0255(美国进入3期临床)和减重药物CG-0416的研发。陈博士拥有22项发明专利,发表过48余篇国际学术期刊论文。
孙木
首席商务官,河络新图生物科技(上海)有限公司
湘雅医学院临床医学学士;吉林大学与德国汉堡大学联合培养生理学博士;复旦大学/美国华盛顿大学EMBA;美国HHMI博士后。
曾先后任职GSK资深科学家,金赛药业商务发展总监,诺华中国合作创新总监,恩华药业首席商务发展官,星奕昂商务发展及战略副总裁,具有丰富的学术界和医药界经验。深耕肿瘤,神经,内分泌等多个疾病领域,迄今在Science等国际著名学术杂志发表20多篇论文。
陈永奇
创始人/董事长/首席科学家,深圳瑞思普利生物制药股份有限公司
陈永奇博士,清华大学硕士、剑桥大学Ph.D,瑞思普利董事长、创始人、首席科学家。陈永奇博士拥有20年以上吸入制剂产品的研发工作经验,曾在英国著名药企VECTURA中任高级技术管理人员、制剂开发部门首席科学家等。在全球已上市的吸入粉雾剂17个产品中,有13个主流品种陈永奇博士均有成功开发经历,包括GSK最新Ellipta系列,并有欧盟,美国成功批准以及成功授权案例。其中陈永奇博士所主导的舒利迭仿制药为目前唯一被欧盟批准上市的仿制药,主导的舒利迭仿制药也在2020年底在美国成功获批。
肩负国产吸入制剂崛起的使命,2018年回国创业,力求早日让国产干粉吸入制剂突破。
大会议程
往期回顾
(点击图片查看第八届APAC 会后报告)
会议亮点
一对一约见系统
通过先进的在线约见系统,我们将为您提供最高效的平台发现并约见目标客户。参会全体人员可选择5~20场商务会前会上预约schedule,20分钟每场,上下午茶歇开始,持续到会议结束,小会议室见面。
明星项目路演
在生物技术专场中我们将邀请在某个领域获得突破性进展并在寻求合作、投资机会的企业参与。鉴于大部分参会者是医药企业中正在寻求合作项目、负责商务拓展、技术许可、合作等部门的高层或是投资人,本环节将是这些生物技术公司展现优势项目、寻找合作伙伴的绝佳舞台。
社交晚宴派对
大会第一天晚上将安排社交晚宴,所有演讲嘉宾及重要嘉宾将被邀请参会晚宴。在轻松、欢快的氛围下与全球医药行业领袖把酒言欢,或许能碰撞出不一样的火花!赶快抓住这次机会!
关键议题
全球化浪潮迭起:中国药企出海的布局与实践
跨越国界,引领创新:全球医药生态的策略与实践
国际资本加持:跨国药企在华投资的考量与选择
全球化升级-MNC的多元化产品组合布局策略
创新“加速度”:多元化分工合作提升药物研发效率
稳健前行:药企全球市场中合规能力的塑造
交易洞察:药企并购重组与商业合作趋势思考
谋时而动,顺势而为:创新药企商业模式革新与策略构建
产学研融合:医药创新生态体系的布局与构建
BD”内循环”:药企本土BD交易新模式的探索
“淘金”热潮前移:早研阶段下的BD交易关键考量
医药“新风向”:New Co模式下的合作与共赢
从资本视角看药企的生存路径思考
资本市场如何挖掘更有价值的创新资产?
关键词概览
END
【咨询参会/媒体合作/赞助合作】
Sunny Sun 孙女士
T: 021 6095 0241
M: 189 6294 0579(微信同号)
添加微信请注明“医药合作峰会”
E-mail: sunny.sun@shine-consultant.com
SHINE CONSULTANT
上海士研管理咨询有限公司成立于2005年,致力于为组织领导者提供沟通交流与专家智库平台。通过二十年沉淀与积累,覆盖了金融与投资、交通与运输、消费与文旅、医药与医疗、能源与资源、高科与电信、公用与政府等产业领域,服务着全球500强和万余家领导型企业,汇聚了百万余名机构决策者,并与千余家产业权威机构建立了战略伙伴关系。士研咨询秉承专业立身的理念发展队伍,现拥有百余名专业的资深人员,核心管理团队都具有十五年以上的专业经验。
寡核苷酸临床申请临床1期
2025-08-10
光鲜背后,是更多的不确定性
近日,一场突然的药品断货潮,让一款此前鲜少被大众提及的肾病药——耐赋康(布地奈德肠溶胶囊)成为市场关注的焦点。
这款由云顶新耀引进并商业化的创新药,是国内首个且目前唯一获批用于治疗IgA肾病(一种慢性肾病)的对因药物。在全国多个城市,出现耐赋康大面积断货、患者排队等药的情况。
小红书等社交平台上,大量患者发帖询问和交流耐赋康断药情况,或收集和交换部分地区耐赋康恢复供货的信息。
焦虑情绪蔓延,反映出中国IgA肾病患者庞大的用药需求长期未被满足。
与耐赋康一同受到关注的,还有其背后的创新药企——云顶新耀。2024年云顶新耀凭借耐赋康在国内大卖首次实现年度商业化盈利,这家三年前还在生死边缘徘徊的公司,如今成为国内创新药企的商业化典范。
仅耐赋康一款药品,年度销售峰值预测就超50亿元,可见云顶新耀商业化前景之广阔。但与此同时,其大股东康桥资本又在大规模减持公司股票。今年1月和6月,康桥资本共有两笔大宗减持,合计超13%。
冰与火交锋中的云顶新耀,将走向何方?
断货背后
肾病蓝海市场的供需失衡
IgA肾病是一种严重的慢性肾脏疾病,患者会出现血尿、蛋白尿,肾功能持续恶化,最终可能发展为尿毒症。
这种疾病好发于青壮年,一旦患病往往意味着要在人生最好的年华里要与病魔长久抗争。
长期以来,医生只能通过降压药等手段延缓病情,缺乏真正的对因治疗药物。耐赋康的出现填补了这一空白,它能够直接作用于疾病根源,阻止病情进一步恶化。2024年5月,该药获得国家药监局完全批准。
耐赋康自上市以来销售表现强劲,并于今年正式被纳入医保,迅速成为市场焦点。根据云顶新耀披露的数据,耐赋康自2024年5月商业化以来,已累计服务超过两万名患者。
未进医保前,耐赋康在2024年仅7个月的销售收入就达到3.53亿元。
进入医保后的放量更为明显,主要是由于价格的大幅下降:耐赋康的单瓶价格由约2.38万元降至5000元左右,部分地区医保报销后患者自付约1000元,极大提升了药物的可负担性和市场渗透率。
业内预测,耐赋康在2025年全年销售额有望突破10亿元,成为罕见的医保首年即达“十亿量级”的慢病创新药,市场表现甚至超出很多传统明星肿瘤药物。
耐赋康的快速放量在于其所处的治疗领域存在高度未满足需求。
在美国,IgA肾病属于罕见病,患者基数仅约13万-15万人。而中国确诊的IgA肾病患者数达到数百万级别,每年新增超过10万人,且亚洲地区患者疾病进展更快、预后更差。
IgA肾病作为慢性肾脏病,难以彻底治愈,多数患者需要长期坚持用药,断药可能导致疾病进展加速,增加发展为终末期肾病的风险。
断货问题的出现与其产能结构密切相关。耐赋康是一款授权引进产品,2019年云顶新耀与瑞典药企Calliditas签订授权许可协议,获得在大中华地区和新加坡开发以及商业化的权利。
目前耐赋康全部依赖境外生产,生产企业为美国公司Patheon Pharmaceuticals。云顶新耀在2024年底时已经开始筹备本地化生产,今年2月向国家药监局药品审评中心递交了扩大产能的补充申请,注册分类为境外上市改良型原研药。
不过通过审评后,由于涉及技术转移、生产验证及质量一致性评估,国内产能难以快速释放,现有海外供应难以完全满足医保落地后的市场需求,耐赋康短期内仍将面临供应不足的问题。
此次断货事件,既展现了肾病治疗领域的巨大市场需求,也让云顶新耀这家以License-in(授权引进药品)模式起家的创新药公司引起了市场关注。
生死逆转
从崩溃边缘到商业化盈利
2025年4月,云顶新耀宣布获香港联交所批准,正式摘掉股票代码中的“B”标记,标志着其脱离“未有盈利”的18A阶段,步入商业化新周期。
公司披露数据显示,2024年全年营收超过7亿元,同比增长461%,毛利率达到83%,非国际财务准则亏损总额同比收窄25%,现金储备达16亿元人民币,运营费用占比显著下降。
成立7年就实现年度商业化盈利,这与其License-in策略密切相关。
License-in模式最关键的是以资本来买时间和确定性,可以略过早期研发阶段,直接授权引进处于研发后期的产品,从而短期内获取更大回报。这让云顶新耀走出了和其他创新药公司不同的道路。
但就在3年前,云顶新耀还在生死边缘挣扎。
2022年,云顶新耀的股价连续下行,市值快速蒸发。公司每股股价从高点104港元一路暴跌至6港元左右。
公司最受市场关注的肿瘤管线戈沙妥珠单抗(拓达维)在当时已进入商业化前夜。该药是云顶新耀在2019年从美国Immunomedics公司引进的ADC(抗体药物偶联物,是一种新型的肿瘤治疗药物)肿瘤产品,是全球首个获批的Trop-2靶点ADC药物。
2020年该产品获得美国药监局(FDA)批准,2022年6月在中国获批上市,成为云顶新耀首款获批的新药,也一度被视为公司市值支撑的重要资产。
然而2022年8月,云顶新耀宣布将拓达维在大中华区、韩国及部分东南亚市场的全部权益转让给吉利德科学,获得总额4.55亿美元的交易对价,其中包括2.8亿美元现金。原先由拓达维构建的肿瘤商业化体系随即消散。
这一决定在当时引发外界广泛关注。一个月后,公司进行全员大会,远在新加坡出差的董事会主席傅唯通过视频宣布,肿瘤团队将整体遣散。
将拓达维剥离后,公司战略方向随之转变。云顶新耀全面收缩肿瘤板块,聚焦于肾病、自体免疫、重症感染等治疗领域,并逐步明确以商业化效率为导向的产品选择机制。
2022年,原CEO卸任,罗永庆出任新任首席执行官,全面接管业务和组织架构调整工作。
不同于上一任CEO擅长研发,罗永庆成功领导过多款药品的临床开发、药政事务和商业化,其商业化能力尤为见长。
罗永庆2016年成为吉利德科学中国总部的首位员工,担任全球副总裁及中国区总经理,在其带领下,吉利德中国团队将多个创新药物引入中国,涉及乙型肝炎、丙型肝炎、艾滋病领域。
2023年伊始,罗永庆开始广泛对外界强调公司在肾病领域的聚焦,其中重点将会推进耐赋康上市,从而奠定云顶新耀在肾科领域的基础,并开发针对肾小球疾病的BTK抑制剂。
但生物医药行业在2022年的普遍低迷,使得云顶新耀这一策略并未让市场太兴奋,不过此后的商业化进程证明了其押注的正确性。
到2024年底,耐赋康已经成为医药市场不折不扣的明星产品,公司也完成销售组织扩容,迎来新的高光时刻。
资本博弈
孵化者兼大股东连续减持
云顶新耀能够迅速调整战略,与其诞生和运作的模式有关。这家公司是康桥资本孵化控股投资策略的典型产物。
康桥资本并非传统财务投资者,而是亚洲最大的医疗健康产业投资运营机构之一,总管理规模为105亿美元。康桥资本已主导和深度参与了云顶新耀、天境生物、苏桥医药、优锐医药、安瑞医疗等多家医疗创新企业的发展。
从2017年创立开始,康桥资本主导了云顶新耀A轮和B轮融资,分别为5000万美元和6000万美元,采用“孵化+运营”模式,从创立初期就深度介入战略规划。
康桥资本在云顶新耀IPO完成前持有62.48%股权,康桥资本首席执行官傅唯本人担任云顶新耀董事会主席。
2019年,康桥资本以8.35亿美元为云顶新耀引入拓达维的亚洲独家权益,此举也推动云顶新耀2020年在港股成功IPO,首日市值突破200亿港元。
而此后云顶新耀抛弃这一资产、解散肿瘤团队,并聚焦肾病等慢病产品的果断动作,同样是康桥资本主导的手笔。
目前市场普遍推测耐赋康未来的年度销售峰值可达50亿元,公司另一款溃疡性结肠炎药物伊曲莫德销售峰值可达20亿元。按照业内估算,云顶新耀引进耐赋康的费用为2.8亿美元,销售分成约10%,整个生命周期预计能赚300亿元。公司商业化和盈利前景一片大好。
7月25日,云顶新耀宣布拟配股融资逾15.7亿港元,配售价69.7港元/股,配售股份占扩大后总股本约6.4%。募集资金中,40%将用于商业化,体现了其对进一步商业化变现的迫切需求。
此次配售价格折让幅度达到10.12%,虽然在港股市场属于常见区间,但考虑到云顶新耀股价处于阶段性高位、基本面向好、市场预期乐观,这一折让幅度仍可能被市场解读为融资方存在一定出手压力,或反映出投资人与公司在估值上的分歧。
且就在前不久的6月,康桥资本作为云顶新耀大股东进行了一轮大宗减持,套现超10亿港元,减持股本比例7.63%。
而在今年1月,康桥资本刚刚减持了1710万股,减持5.2%。两次减持动作合计减持股份约13%,目前康桥资本持股比例已降至25.92%。
一边是明星产品供不应求,一边是大股东在真正的商业化兑现前逐步抽身。这种时间差值得关注。康桥资本给出的理由是“国际知名投资机构的入市兴趣促使适时出售”,以及“进一步优化投资人结构”。
但作为私募股权基金,康桥资本面临LP(有限合伙人)的退出需求,尤其是在云顶新耀股价从低位反弹超237%后,减持确实能实现丰厚收益并缓解基金流动性压力。
但对这家估值高度依赖产品转化效率的公司来说,大股东的节奏变化难免引发外界对后续路径的更多揣测。
目前,云顶新耀已形成相对完整的产品矩阵。商业化产品包括耐赋康、依嘉、维适平三款,覆盖肾病、抗感染、自免疾病领域。
在研管线中,BTK抑制剂EVER001在膜性肾病治疗中展现出亮眼数据,有望年内落地海外授权合作。此外,公司也在积极发展mRNA、CAR-T等细胞与基因治疗技术平台。
云顶新耀的未来能否持续闪耀?我们将持续关注。
【来源】子弹财经
【作者】张钰
【编辑】胡芳洁
免责声明(上下滑动查看全部)
任何在本文出现的信息(包括但不限于个股、评论、预测、图表、指标、理论、任何形式的表述等)均只作为参考,投资人须对任何自主决定的投资行为负责。另,本文中的任何观点、分析及预测不构成对阅读者任何形式的投资建议,亦不对因使用本文内容所引发的直接或间接损失负任何责任。投资有风险,过往业绩不预示未来表现。财经下午茶力求文章所载内容及观点客观公正,但不保证其准确性、完整性、及时性等。本文仅代表作者本人观点。
基因疗法上市批准核酸药物引进/卖出
2025-07-23
·药时空
7月21日,北京康辰药业股份有限公司全资子公司康辰药业(香港)有限公司(“康辰香港”)与优锐开曼及其下属子公司共同签署了《Series EPreferred Share Purchase Agreement》(“《股份认购协议》”),康辰香港拟以1.5亿元人民币价格,参与优锐开曼E轮融资,认购优锐开曼新发行的38,439,537 股E轮优先股,此外北京市医药健康产业投资基金(有限合伙)及北京昌平产业发展投资基金(有限合伙)也以1.5亿元参与认购38,439,537股E轮优先股。本轮融资募集资金将用于优锐开曼业务拓展、资本支出与营运资金等,以支持其创新药申报上市,以及满足其他经营资金需求。优锐医药由康桥资本孵化的创新药引进和商业化分销的CSO公司。优锐开曼下属子公司优锐生物医药(北京)有限公司因现有产品本地化生产向公司租赁厂房等生产设施,租期5年,租金共计7500万元。该租金中的5000万元折合为康辰香港对优锐开曼的投资款,专项认购优锐开曼新发行的E轮优先股。同时,为强化双方的战略合作,康辰药业以自有资金再行增加投资1亿元通过康辰香港认购优锐开曼新发行的E轮优先股(即两项合计金额1.5亿元人民币)。本次增资完成后,康辰药业向优锐开曼累计投资金额为31,203.25万元人民币;康辰药业通过康辰香港将持有优锐开曼91,448,871股股份,占优锐开曼总股本(全面摊薄和转股基础上)的比例已超过达到7.98%。优锐开曼设立于开曼群岛,通过全资子公司Nuance Pharma Limited及二级 全资子公司优锐医药科技(上海)有限公司(“优锐医药”)开展实际运营。优锐医药是一家以患者为中心、聚焦创新的生物制药公司,拥有呼吸类创新研发管线与成熟的商业化产品组合,专注于呼吸系统、缺铁性贫血等治疗领域。其当前重点在研管线恩司芬群(Ensifentrine),是一款全球首创的吸入型磷酸二酯酶3、4双靶点抑制剂(PDE3/PDE4),用于维持治疗慢性阻塞性肺疾病 (COPD)。该药物凭借双重抑制机理,使其能够凭借单个化合物同时实现支气管扩张以及抗炎效应,已于2024年在美国获批上市(合作伙伴Verona Pharma已经被默沙东收购),并获批落地中国海南博鳌乐城先行区;2025年,该药在中国澳门特别行政区获批上市,同年5月在中国大陆COPD三期临床试验中达到主要终点,计划下半年提交新药申请。截至2025年3月31日,优锐开曼最近一年及一期合并报表主要财务数据如下:优锐开曼是一家高度整合的、聚焦特定治疗领域的创新型生物医药公司,拥有领先的商务和临床团队,在海外创新药、原研药引入等方面具有业内公认的资源渠道优势,其核心管理人员及销售团队具有多年进口药品在中国国内成功的商业运营经验。本次投资完成后,康辰药业也将借助优锐开曼的业务平台和业务资源,拓展公司创新药及原研药国际合作的商业化运营,助力公司战略目标的实现。识别微信二维码,可添加药时空小编请注明:姓名+研究方向!
临床3期并购申请上市
100 项与 优锐医药科技(上海)有限公司 相关的药物交易
登录后查看更多信息
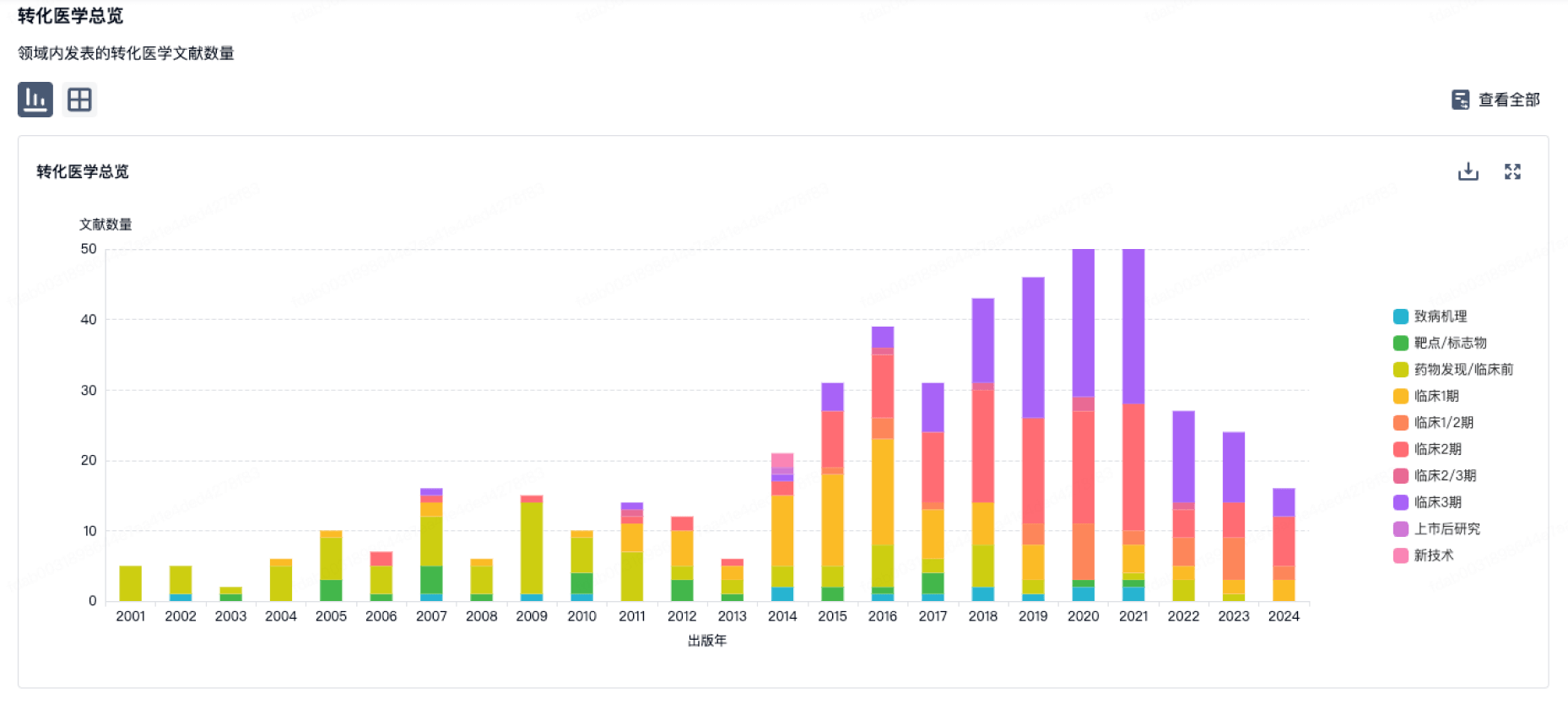
100 项与 优锐医药科技(上海)有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年11月07日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
申请上市
1
1
批准上市
其他
2
登录后查看更多信息
当前项目
| 药物(靶点) | 适应症 | 全球最高研发状态 |
|---|---|---|
恩塞芬汀 ( PDE3 x PDE4 ) | 慢性阻塞性肺疾病 更多 | 批准上市 |
MVA-BN RSV(Bavarian Nordic) | 呼吸道合胞体病毒感染 更多 | 终止 |
登录后查看更多信息
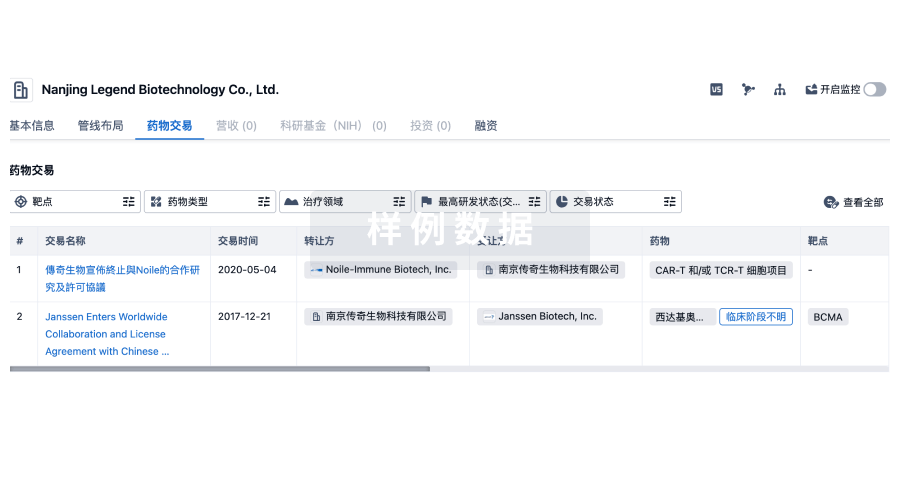
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
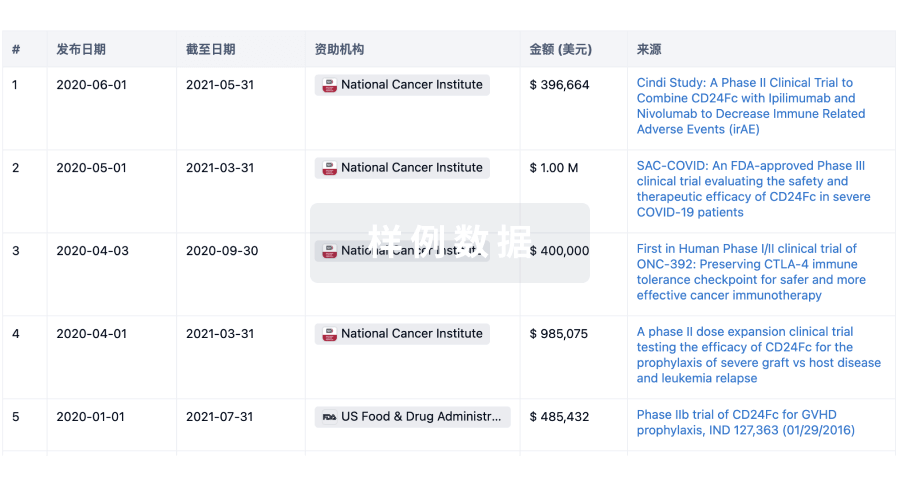
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用