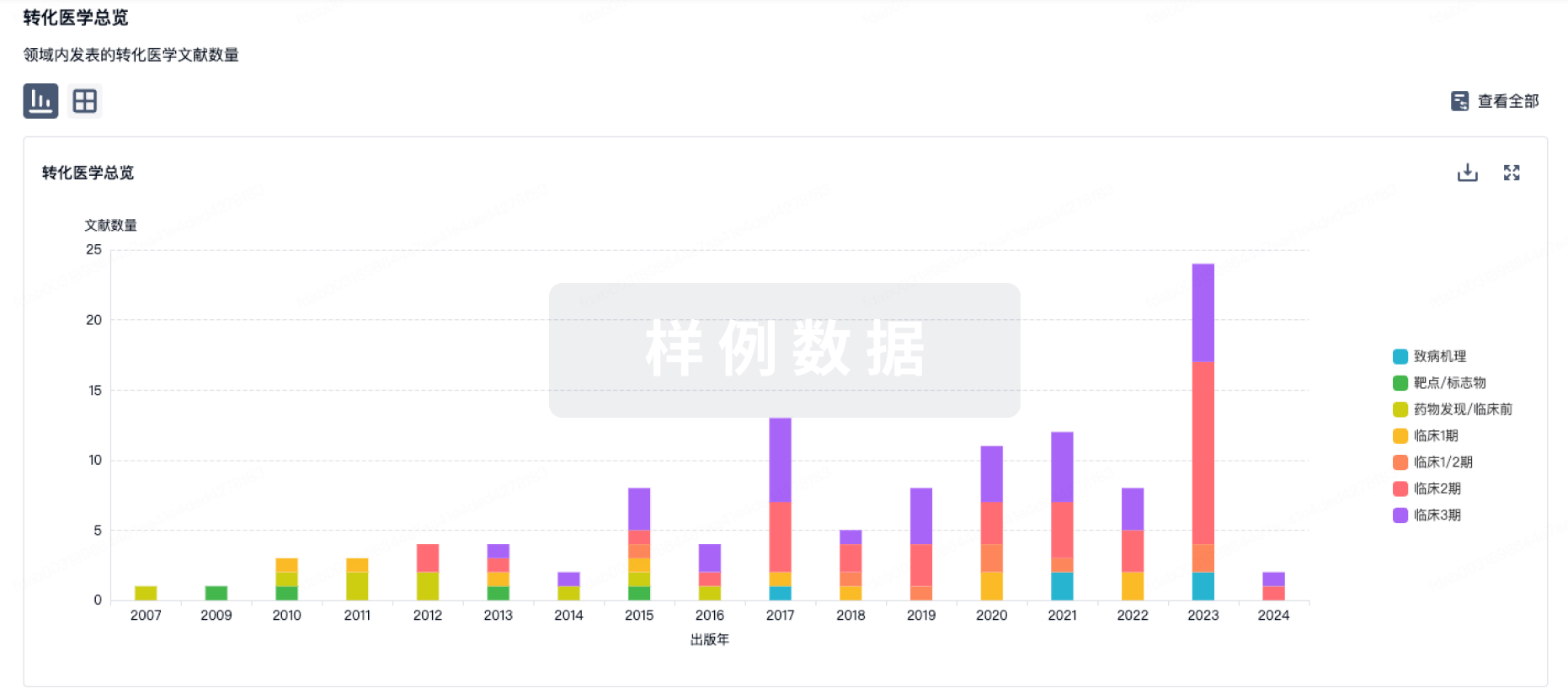
本周药品注册受理数据,分门别类呈现,一目了然。(5.26-6.1)新药上市申请药品名称企业注册分类受理号伊托法替布软膏明慧医药(杭州)有限公司1CXHS2500058洛布替尼片广州麓鹏制药有限公司1CXHS2500057洛布替尼片广州麓鹏制药有限公司1CXHS2500056ACYW???群脑膜炎球菌多糖结合疫苗北京智飞绿竹生物制药有限公司3.2CXSS2500056注射用STSP-0601江苏贝捷泰生物科技有限公司1CXSS2500057重组人源化单抗MIL62注射液北京天广实生物技术股份有限公司1CXSS2500054新药临床申请药品名称企业注册分类受理号BGB-16673片百济神州(苏州)生物科技有限公司1CXHL2500526BGB-16673片百济神州(苏州)生物科技有限公司1CXHL2500525HSK47388片海思科医药集团股份有限公司1CXHL2500521SZ2001片苏中药业集团股份有限公司1CXHL2500524SZ2001片苏中药业集团股份有限公司1CXHL2500523HSK47388片海思科医药集团股份有限公司1CXHL2500522RO7840734罗氏(中国)投资有限公司1CXHL2500519RO7840734罗氏(中国)投资有限公司1CXHL2500518SYN818片杭州圣域生物医药科技有限公司1CXHL2500511SYN818片杭州圣域生物医药科技有限公司1CXHL2500510富马酸奥比特嗪肠溶微丸胶囊深圳市真兴医药技术有限公司1CXHL2500512HEC20002胶囊广东东阳光药业股份有限公司2.2CXHL2500527CZ006注射液浙江萃泽医药科技有限公司2.2CXHL2500520硫酸阿托品滴眼液湖南迈欧医疗科技有限公司2.2CXHL2500516KEM2417缓释干混悬剂深圳珐玛易药品科技有限公司2.2CXHL2500515KEM2417缓释干混悬剂深圳珐玛易药品科技有限公司2.2CXHL2500514硫酸阿托品滴眼液湖南迈欧医疗科技有限公司2.2CXHL2500517MZ-01吸入溶液成都米子生物医药科技有限公司2.2;2.4CXHL2500513鼻喷重组呼吸道合胞病毒疫苗(5型副流感病毒载体)广州思安信生物技术有限公司1.1CXSL2500430重组呼吸道合胞病毒疫苗(CHO细胞)(佐剂)江苏中慧元通生物科技股份有限公司1.2CXSL2500429三价流感病毒裂解疫苗(MDCK细胞)兰州百灵生物技术有限公司2.2CXSL2500428注射用YL201苏州宜联生物医药有限公司1CXSL2500441注射用BAT8008百奥泰生物制药股份有限公司1CXSL2500440BAT1308注射液百奥泰生物制药股份有限公司1CXSL2500438SM2275注射液珠海烁星生物医药科技有限公司1CXSL2500443注射用YL202苏州宜联生物医药有限公司1CXSL2500437注射用HB0043上海华奥泰生物药业股份有限公司1CXSL2500431注射用YL201苏州宜联生物医药有限公司1CXSL2500436IBI363信达生物制药(苏州)有限公司1CXSL2500435IBI363信达生物制药(苏州)有限公司1CXSL2500434注射用MHB088C明慧医药(杭州)有限公司1CXSL2500433注射用MHB039A明慧医药(杭州)有限公司1CXSL2500432HEC-301注射液东莞市东阳光生物药研发有限公司1CXSL2500426注射用BL-B01D1成都百利多特生物药业有限责任公司1CXSL2500424重组人PD-L1/CD3 双抗溶瘤II型单纯疱疹病毒注射液(Vero细胞)武汉滨会生物科技股份有限公司1CXSL2500425注射用RMMV疫苗长春康悦生物科技有限公司1CXSL2500423注射用IMD303亲合力(成都)药业有限公司1CXSL2500422BAT4706注射液百奥泰生物制药股份有限公司2.4CXSL2500439注射用重组A型肉毒毒素乐普健糖药业(重庆)有限公司2.4CXSL2500427仿制药申请药品名称企业注册分类受理号利伐沙班颗粒广州大光制药有限公司3CYHS2501949利伐沙班颗粒广州大光制药有限公司3CYHS2501948硫酸沙丁胺醇注射液亚邦医药股份有限公司3CYHS2501947氟比洛芬贴剂湖南九典制药股份有限公司3CYHS2501944硫酸妥布霉素注射液湖北华鸿医药有限公司3CYHS2501943辅酶Q10片海南元盈医药科技有限公司3CYHS2501942维生素B6注射液上海禾丰制药有限公司3CYHS2501936复方氨基酸注射液(20AA-Ⅱ)内蒙古白医制药股份有限公司3CYHS2501930复方氨基酸注射液(20AA-Ⅱ)内蒙古白医制药股份有限公司3CYHS2501929维生素B6注射液南京海鲸药业股份有限公司3CYHS2501927盐酸氨溴索滴剂浙江赛默制药有限公司3CYHS2501926盐酸氨溴索滴剂浙江赛默制药有限公司3CYHS2501925富马酸酮替芬口服溶液安徽茂康药业有限公司3CYHS2501921中性腹膜透析液(碳酸氢盐-G4.25%)石家庄四药有限公司3CYHS2501913注射用盐酸头孢替安/氯化钠注射液湖南科伦制药有限公司3CYHS2501904盐酸昂丹司琼片南京海鲸药业股份有限公司3CYHS2501901盐酸昂丹司琼片南京海鲸药业股份有限公司3CYHS2501900盐酸普萘洛尔注射液安徽美来药业股份有限公司3CYHS2501895氨磺必利口服溶液福安药业集团湖北人民制药有限公司3CYHS2501908利伐沙班口腔崩解片山东新时代药业有限公司3CYHS2501884利伐沙班口腔崩解片山东新时代药业有限公司3CYHS2501883奥美拉唑碳酸氢钠干混悬剂(II)裕松源药业有限公司3CYHS2501882复方聚乙二醇电解质散(II)湖南润兴药业有限公司3CYHS2501876腺苷钴胺胶囊济南中渔润医药科技有限公司3CYHS2501874乙酰半胱氨酸注射液长春澜江医药科技有限公司4CYHS2501957洛索洛芬钠贴剂津药达仁堂集团股份有限公司新新制药厂4CYHS2501956洛索洛芬钠贴剂津药达仁堂集团股份有限公司新新制药厂4CYHS2501955巴瑞替尼片广州大光制药有限公司4CYHS2501954脂肪乳氨基酸(17)葡萄糖(19)注射液瑞阳制药股份有限公司4CYHS2501953盐酸尼卡地平注射液赤峰源生药业有限公司4CYHS2501952盐酸尼卡地平注射液赤峰源生药业有限公司4CYHS2501951乙酰半胱氨酸注射液湖北津药药业股份有限公司4CYHS2501950氟伐他汀钠缓释片浙江百代医药科技有限公司4CYHS2501946乙酰半胱氨酸注射液合肥启旸生物医药科技有限公司4CYHS2501945聚乙烯醇滴眼液湖南华纳大药厂股份有限公司4CYHS2501941环索奈德吸入气雾剂上海上药信谊药厂有限公司4CYHS2501937硫酸特布他林雾化吸入用溶液上海旭东海普药业有限公司4CYHS2501938注射用伏立康唑湖北午时医药研究院有限公司4CYHS2501935瑞巴派特片浙江浙北药业有限公司4CYHS2501934磷酸西格列汀片云鹏医药集团有限公司4CYHS2501933西格列汀二甲双胍片(II)江西施美药业股份有限公司4CYHS2501932达可替尼片北京康立生医药技术开发有限公司4CYHS2501931依达拉奉右莰醇注射用浓溶液陕西丽彩药业有限公司4CYHS2501928恩他卡朋双多巴片(II)复星万邦(江苏)医药集团有限公司4CYHS2501940盐酸乙哌立松片西洲医药科技(浙江)有限公司4CYHS2501939玻璃酸钠滴眼液润尔眼科药物(广州)有限公司4CYHS2501918蒙脱石散成都恒瑞制药有限公司4CYHS2501916比拉斯汀片新乡市常乐制药有限责任公司4CYHS2501914乌帕替尼缓释片广东华润顺峰药业有限公司4CYHS2501912非诺贝特酸胆碱缓释胶囊苏州特瑞药业股份有限公司4CYHS2501911氟比洛芬钠滴眼液广州大光制药有限公司4CYHS2501910布瑞哌唑片杭州和康药业有限公司4CYHS2501924布瑞哌唑片杭州和康药业有限公司4CYHS2501923布瑞哌唑片杭州和康药业有限公司4CYHS2501922乙酰半胱氨酸注射液浙江赛默制药有限公司4CYHS2501920乙酰半胱氨酸注射液四川科伦药业股份有限公司4CYHS2501919乙酰半胱氨酸注射液广州大光制药有限公司4CYHS2501917乙酰半胱氨酸注射液广州君博医药科技有限公司4CYHS2501915克林霉素磷酸酯过氧苯甲酰凝胶江苏知原药业股份有限公司4CYHS2501905依折麦布阿托伐他汀钙片(II)山东创新药物研发有限公司4CYHS2501903依折麦布阿托伐他汀钙片(I)山东创新药物研发有限公司4CYHS2501902艾普拉唑肠溶片江苏和晨药业有限公司4CYHS2501899结构脂肪乳注射液(C6-24)成都国为生物医药有限公司4CYHS2501898盐酸左布比卡因注射液河南普瑞药业有限公司4CYHS2501897盐酸左布比卡因注射液河南普瑞药业有限公司4CYHS2501896硫酸氨基葡萄糖胶囊福州闽海药业有限公司4CYHS2501894左甲状腺素钠片江苏德源药业股份有限公司4CYHS2501893左甲状腺素钠片江苏德源药业股份有限公司4CYHS2501892氧云县长隆工业气体有限公司4CYHS2501891达格列净片陕西君可力医药科技有限公司4CYHS2501890达格列净片陕西君可力医药科技有限公司4CYHS2501889硫酸氨基葡萄糖胶囊唐山利康药业有限责任公司4CYHS2501888盐酸阿罗洛尔片长春海悦药业股份有限公司4CYHS2501907克立硼罗软膏山东达冠医药科技有限公司4CYHS2501906盐酸尼卡地平注射液湖南赛隆药业(长沙)有限公司4CYHS2501909地夸磷索钠滴眼液海南斯达制药有限公司4CYHS2501887阿达帕林过氧苯甲酰凝胶江苏知原药业股份有限公司4CYHS2501886阿达帕林过氧苯甲酰凝胶江苏知原药业股份有限公司4CYHS2501885氯吡格雷阿司匹林片湖南尚众合生物医药有限公司4CYHS2501881苹果酸奈诺沙星氯化钠注射液广州大光制药有限公司4CYHS2501880阿达帕林凝胶乐泰药业(兰西)有限公司4CYHS2501879酒石酸美托洛尔片山东普瑞曼药业有限公司4CYHS2501878酒石酸美托洛尔片山东普瑞曼药业有限公司4CYHS2501877阿司匹林肠溶片赤峰万泽药业股份有限公司4CYHS2501875雷珠单抗注射液杭州中美华东制药有限公司3.3CXSS2500055硫酸特布他林注射液广东万泰科创药业有限公司3CYHL2500102氨苯砜凝胶深圳珐玛易药品科技有限公司3CYHL2500101去氧胆酸注射液江苏润恒制药有限公司3CYHL2500099卡麦角林片长春金赛药业有限责任公司3CYHL2500098倍氯米松福莫特罗吸入气雾剂鲁南贝特制药有限公司4CYHL2500100冻干b型流感嗜血杆菌结合疫苗艾美坚持生物制药有限公司3.3CXSL2500442进口申请药品名称企业注册分类受理号甲苯磺酸纳地美定片Shionogi & Co., Ltd.5.1JXHS2500060伊奈利珠单抗注射液Horizon Therapeutics Ireland DAC2.2JXSS2500070注射用维泊妥珠单抗Roche Pharma (Schweiz) AG2.2JXSS2500069注射用维泊妥珠单抗Roche Pharma (Schweiz) AG2.2JXSS2500068比奇珠单抗注射液UCB Pharma S.A.3.1JXSS2500067比奇珠单抗注射液UCB Pharma S.A.3.1JXSS2500066比奇珠单抗注射液UCB Pharma S.A.3.1JXSS2500065比奇珠单抗注射液UCB Pharma S.A.3.1JXSS2500064恩他卡朋片Aurobindo Pharma Limited5.2JYHS2500024GIREDESTRANTGenentech, Inc.1JXHL2500122PF-08046047Pfizer Inc.1JXSL2500085Mosunetuzumab Injection (Subcutaneous Injection)F. Hoffmann-La Roche Ltd.2.1JXSL2500084Mosunetuzumab Injection (Subcutaneous Injection)F. Hoffmann-La Roche Ltd.2.1JXSL2500083中药相关申请药品名称企业注册分类受理号桃红四物汤颗粒瑞阳制药股份有限公司3.1CXZS2500023健脾疏肝固本颗粒江苏康缘药业股份有限公司1.1CXZL2500032JMZ-2102胶囊江西济民可信药业有限公司1.2CXZL2500034JMZ-2102江西济民可信药业有限公司1.2CXZL2500033注:绿色字体部分为潜在首仿品种;不包含原料药、医用氧、注射用水、氯化钠或葡萄糖注射液等申请,不包含再注册、一次性进口、技术转移、复审申请。