预约演示
更新于:2025-05-07
Developmental Disabilities
发育障碍
更新于:2025-05-07
基本信息
别名 Child Development Deviation、Child Development Deviations、Child Development Disorder + [45] |
简介 Disorders in which there is a delay in development based on that expected for a given age level or stage of development. These impairments or disabilities originate before age 18, may be expected to continue indefinitely, and constitute a substantial impairment. Biological and nonbiological factors are involved in these disorders. (From American Psychiatric Glossary, 6th ed) |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
ACTRN12624001244594
Impact of CP-KASP (Cerebral Palsy Knowledge, Advocacy Skills, and Support Program) on parenting stress, empowerment and efficacy, and quality of life in caregivers of children with cerebral palsy.

ACTRN12625000363482
Kids Connect Integrated Hybrid program: a tiered care approach to optimise access to screening and support for child’s developmental, parental mental health, and family psychosocial needs for ALL families (including priority populations).
ACTRN12625000339459
Evaluating the safety and clinical effectiveness of the M-Finity stem in patients undergoing total hip arthroplasty through a CT-based migration analysis.
100 项与 发育障碍 相关的临床结果
登录后查看更多信息
100 项与 发育障碍 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2026-01-01Neural Regeneration Research
GEMIN5 and neurodevelopmental diseases: From functional insights to disease perception
Article
作者: Francisco-Velilla, Rosario ; Martinez-Salas, Encarnacion
2025-12-31Future Science OA
Validation of salivary proteomic biomarkers for early detection of oral cancer in the Egyptian population
Article
作者: Ghalwash, Dalia ; Ammar, Ahmed ; Abou-Bakr, Asmaa ; El-Gawish, Ayman ; Diab, Al-Hassan
2025-12-31Expert Review of Vaccines
Immunogenicity and safety of a live attenuated varicella vaccine in healthy subjects aged between 13 to 55 years: a double-blind, randomized, active-controlled phase III clinical trial in China
Article
作者: Sun, Jinfang ; Zhang, Hao ; Zhang, Yang ; Pan, Hongxing ; Li, Jingxin ; Zhu, Fengcai ; Shi, Jinhui ; Chang, Xianyun ; Li, Guifan ; Wang, Shiyuan
2025-05-01
·梅斯医学
临床研究申请上市
分析
对领域进行一次全面的分析。
登录
或
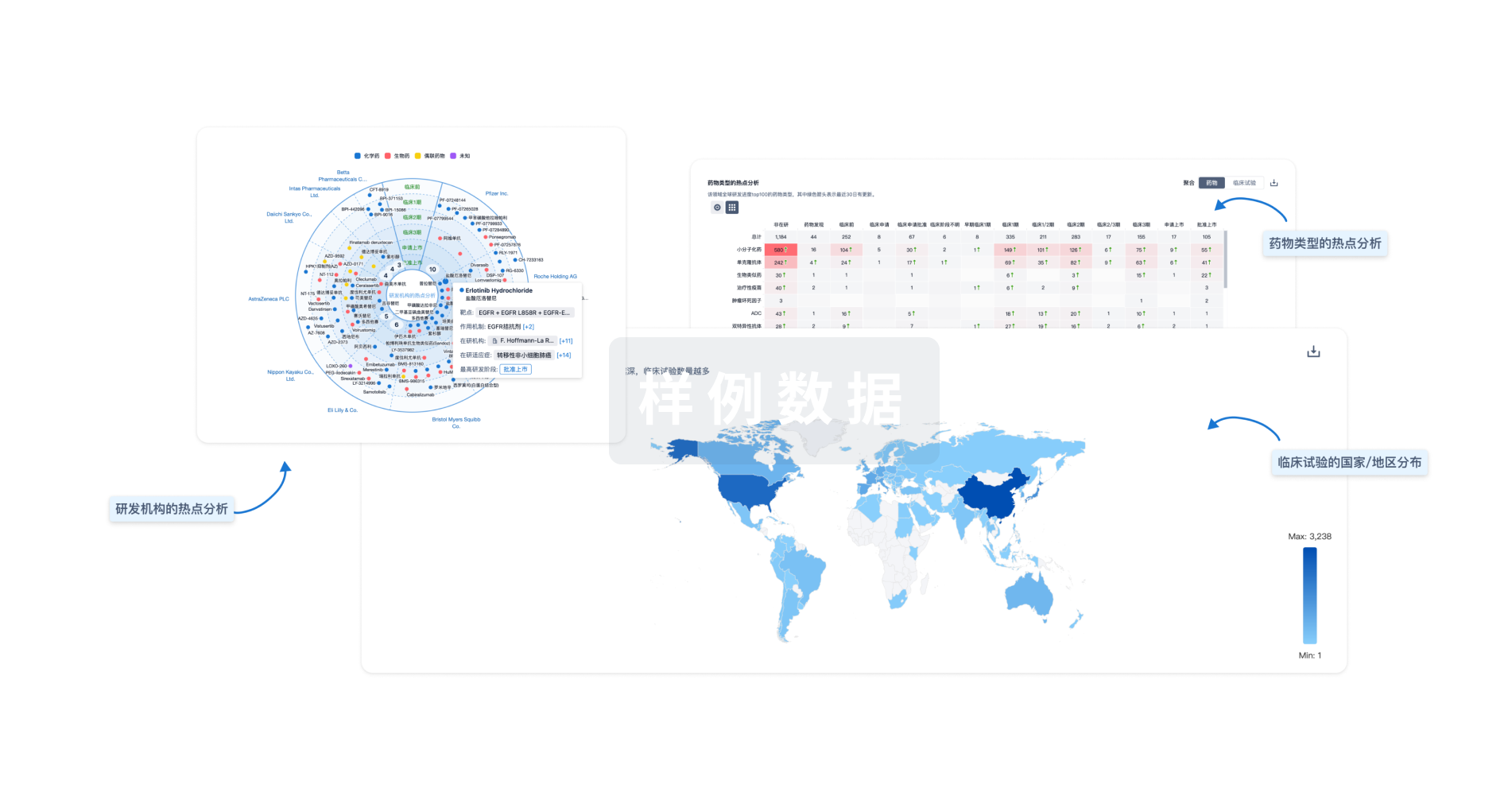
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用