预约演示
更新于:2025-05-07
Primary Parkinson's disease
原发性帕金森病
更新于:2025-05-07
基本信息
别名- |
简介- |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

CTR20250991
甲磺酸溴隐亭片在健康受试者中随机、开放、单剂量、四周期、两序列、完全重复交叉的餐后状态下生物等效性试验
NCT06888869
The Development and Verification of an Interactive Home-based Rehabilitation Exercise Assessment Platform and Exploration of Technology Acceptance and Clinical Effects in Patients with Parkinson's Disease
CTR20244974
UX-DA001注射液(人中脑多巴胺能神经前体细胞注射液)在原发性帕金森病患者中的探索性临床试验
100 项与 原发性帕金森病 相关的临床结果
登录后查看更多信息
100 项与 原发性帕金森病 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2026-01-01Neural Regeneration Research
Adeno-associated viral vectors for modeling Parkinson’s disease in non-human primates
Article
作者: Lanciego, José L. ; Chocarro, Julia
2024-12-01Clinical Genitourinary Cancer
Appearance of New Lesions Associate With Poor Prognosis in Pembrolizumab-Treated Urothelial Carcinoma
Article
作者: Teishima, Jun ; Okamura, Yasuyoshi ; Nakano, Yuzo ; Bando, Yukari ; Hyodo, Yoji ; Hara, Takuto ; Terakawa, Tomoaki ; Suzuki, Kotaro ; Chiba, Koji ; Miyake, Hideaki
2024-10-30Brain Communications
Brain age in genetic and idiopathic Parkinson's disease
Article
作者: Storch, Alexander ; Schumacher, Julia ; Hermann, Andreas ; Hoffmann, Hauke ; Dyrba, Martin ; Teipel, Stefan J
分析
对领域进行一次全面的分析。
登录
或
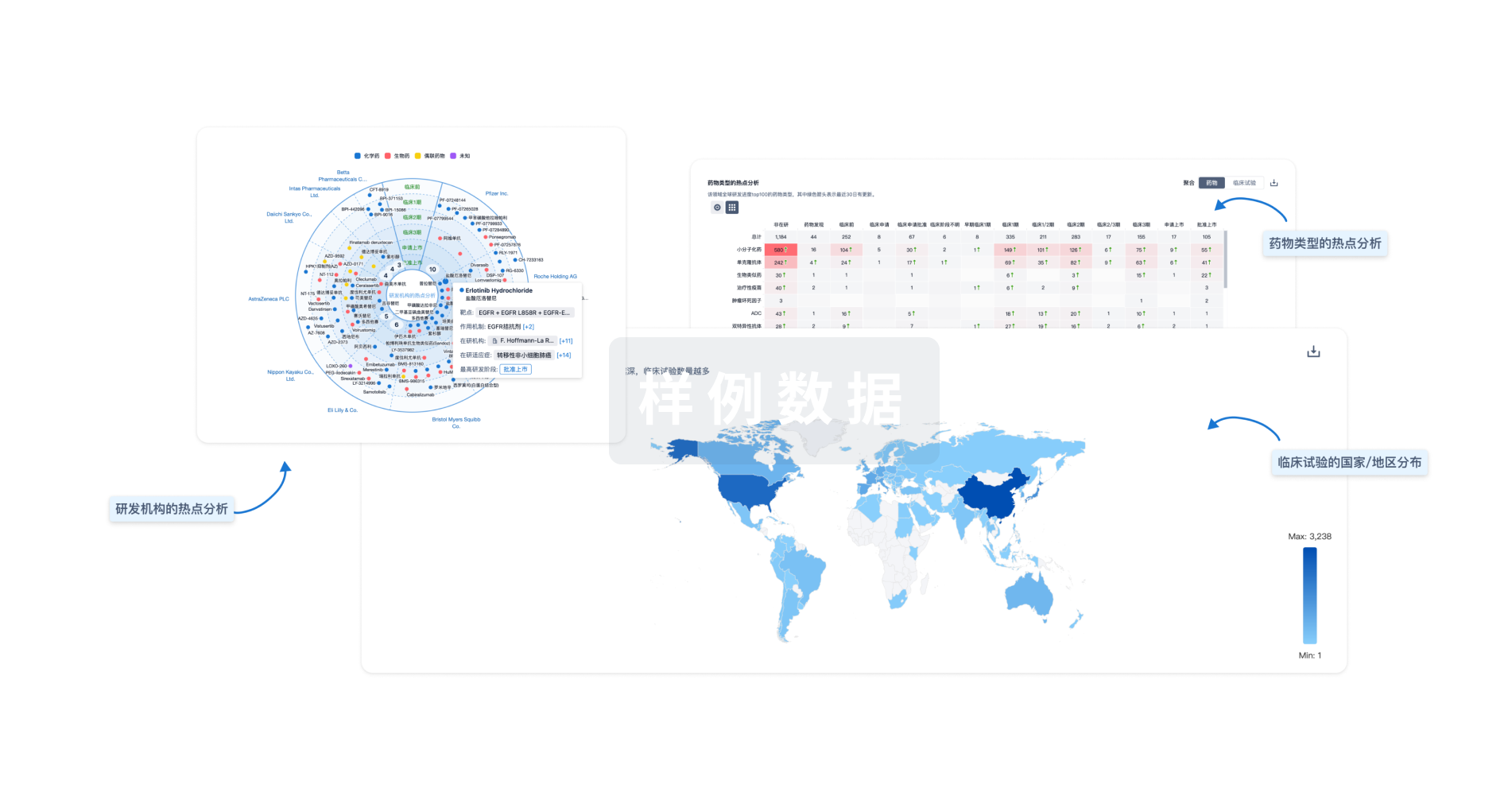
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用