礼来IL-23单抗mirikizumab拟纳入突破性治疗品种
2023年7月24日,中国国家药监局药品审评中心(CDE)官网最新公示,礼来(Eli Lilly and Company)申请的mirikizumab注射液拟纳入突破性治疗品种,拟开发用于治疗中重度活动性克罗恩病(CD)成人患者。
Mirikizumab由礼来公司开发,是一种针对 IL-23 的 p19 亚基的单克隆抗体。2023年3月27日,该药物首次获得日本厚生劳动省(MHLW)批准上市,作为中度至重度活动期溃疡性结肠炎成人的首创治疗药物。此外,欧洲药品管理局(EMA)人用药品委员会(CHMP)对mirikizumab作为对常规治疗或生物治疗应答不足、应答消失或不耐受的中度至重度活动期溃疡性结肠炎成年患者的首选治疗方案发表了积极意见。欧盟委员会于2023年5月26日授予mirikizumab的上市许可。今年6月底,基于LUCENT-1和LUCENT-2III期试验的成功结果,mirikizumab在英国成功获批。
2023年4月13日,礼来公司宣布FDA已就用于治疗溃疡性结肠炎(UC)的Mirikizumab生物许可申请(BLA)发出完整回复函,FDA因生产制造问题拒绝礼来新型抗炎药Mirikizumab上市。在信中,FDA对Mirikizumab的生产制造过程存在疑问,但认可临床数据、安全性和说明书。礼来表示对Mirikizumab的关键3期临床数据及其帮助溃疡性结肠炎患者的潜力充满信心,并将继续与FDA积极合作,争取尽快在美国上市。
此前,mirikizumab已在治疗中重度克罗恩病的2期临床试验中获得积极结果。这是一项名为SERENITY的随机、双盲、含安慰剂对照试验,主要终点是12周之后内镜缓解率,定义为克罗恩病简化内镜评分(SES-CD)与基线相比降低50%。次要终点包括患者报告结局(PRO)检测的临床缓解,以及安全性。试验达到了主要终点和关键次要终点,与安慰剂相比,mirikizumab在治疗12周后显著降低临床和内镜检测到的疾病活动。在内镜缓解率方面,接受200mg、600mg和1000mg剂量mirikizumab的患者组,缓解率分别为25.8%、37.5%和43.8%,安慰剂组这一数值为10.9%。在PRO临床缓解率方面,接受200mg、600mg和1000mg剂量mirikizumab的患者组,缓解率分别为12.9%、28.1%和21.9%,安慰剂组这一数值为6.3%。
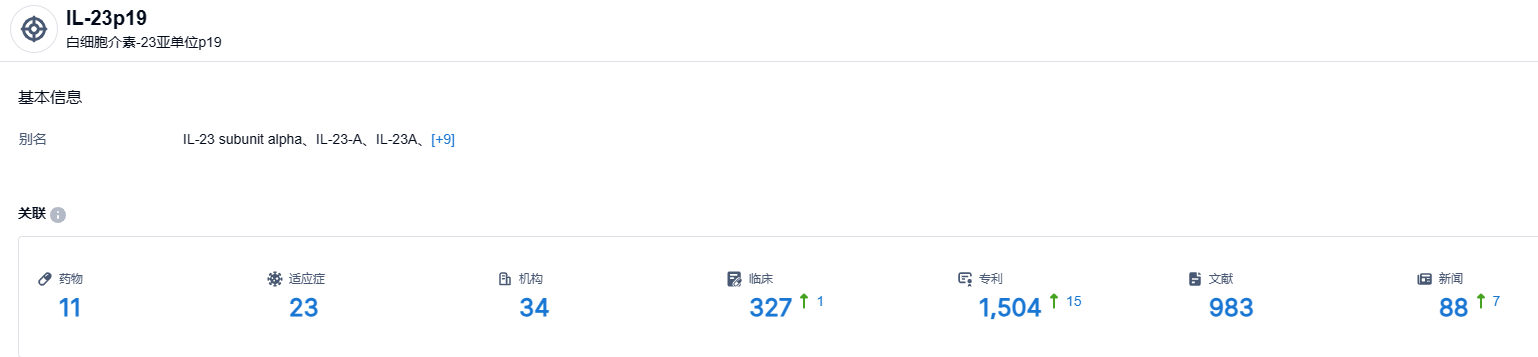
根据智慧芽新药情报库所披露的信息 (点击下方卡片直达IL-23p19 靶点注册登录后可免费获得该靶点下的在研药物、适应症、研发机构、临床试验等详细信息),截止到 2023 年7月25日,IL-23p19靶点共有在研药物11个,包含的适应症有23种,在研机构34家,涉及相关的临床试验327件,专利多达1504件。期待mirikizumab的后续表现。




