预约演示
更新于:2025-05-07
Opioid receptors x μ opioid receptor
更新于:2025-05-07
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

NCT06251609
Naloxone for Opioid Associated Out of Hospital Cardiac Arrest

NCT06854029
Optimizing Long-Acting Pre-Exposure Prophylaxis and Medications for Opioid Use Disorder Interventions in Carceral Settings

100 项与 Opioid receptors x μ opioid receptor 相关的临床结果
登录后查看更多信息
100 项与 Opioid receptors x μ opioid receptor 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-09-01Journal of Colloid and Interface Science
Self-supported copper-cobalt oxide hybrid electrode for bifunctionally electrocatalytic nitrate reduction and methanol oxidation reactions
Article
作者: Gao, Zhiyong ; Ren, Junhao ; Wang, Xiuge
2025-08-01Neuropharmacology
A putative binding model of nitazene derivatives at the μ-opioid receptor
Article
作者: Clayton, Joseph ; Shen, Jana ; Skiniotis, Georgios ; Shi, Lei ; Robertson, Michael J ; Stavitskaya, Lidiya ; Michaelides, Michael
2025-08-01Neuropharmacology
Behavioral economics of polysubstance use: The role of orexin-1 receptors in nicotine-induced augmentation of synthetic opioid consumption
Article
作者: Gilles-Thomas, Elizabeth A ; McSain, Shannon L ; Honeycutt, Sarah C ; Loney, Gregory C ; Mukherjee, Ashmita ; Lichte, David D
2025-04-27
抗体药物偶联物财报蛋白降解靶向嵌合体多肽偶联药物核酸药物
2025-03-14
上市批准临床3期临床结果
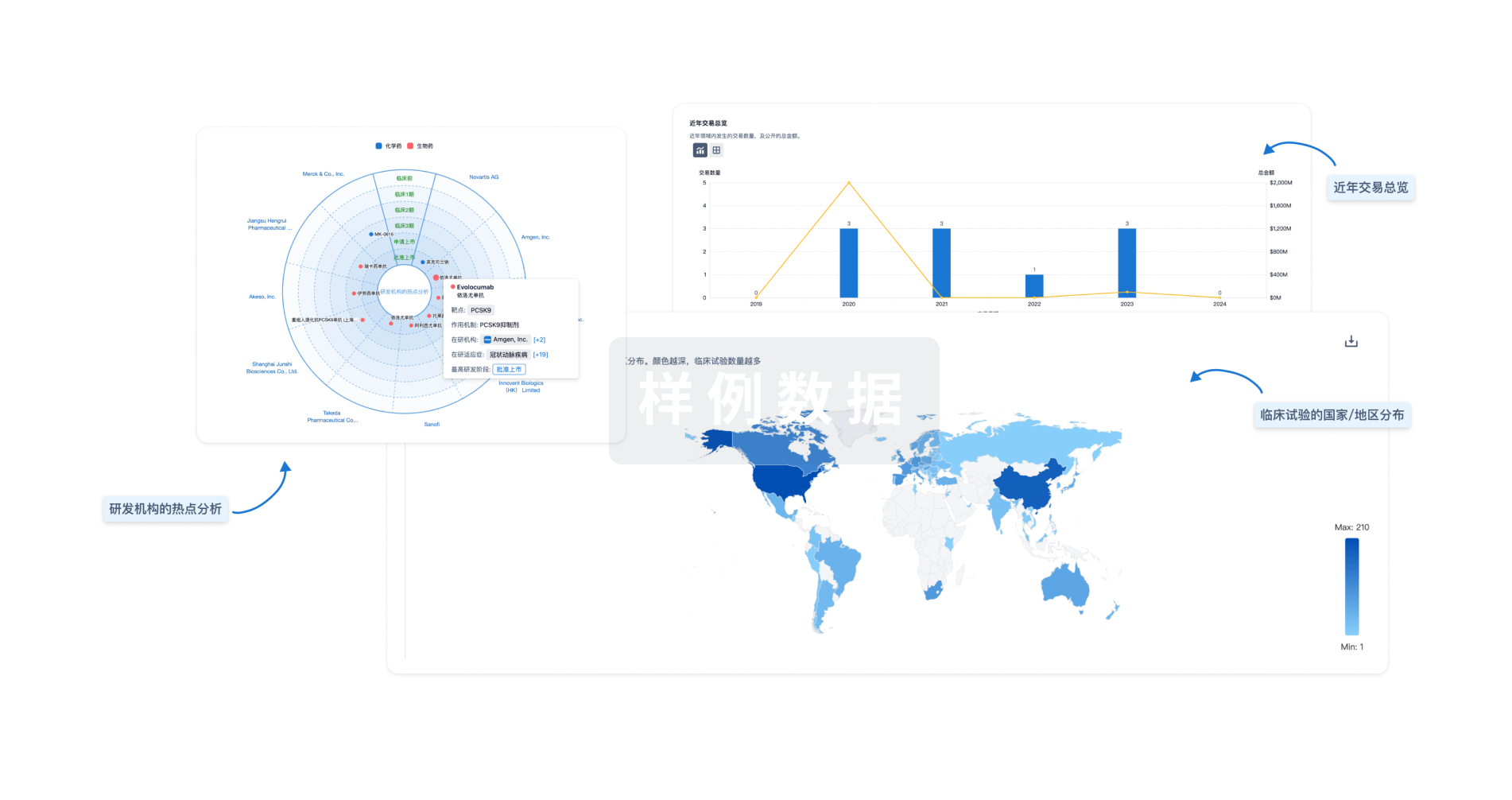
分析
对领域进行一次全面的分析。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用