预约演示
更新于:2025-08-29

Cassiopea SpA
更新于:2025-08-29
概览
标签
皮肤和肌肉骨骼疾病
小分子化药

关联
1
项与 Cassiopea SpA 相关的药物靶点 |
作用机制 AR拮抗剂 |
在研机构 |
非在研适应症- |
最高研发阶段批准上市 |
首次获批国家/地区 美国 |
首次获批日期2020-08-26 |
7
项与 Cassiopea SpA 相关的临床试验NCT05914805
A 6-month Phase 3, Multicenter, Prospective, Randomized, Double-Blind, Vehicle-Controlled Study, to Evaluate the Efficacy and Safety of Topically Applied Clascoterone Solution for the Treatment of Androgenetic Alopecia in Males, Followed by a 6-month Single-Blind Treatment With Clascoterone or Vehicle Solution
The purpose of the study is to see if Clascoterone can help people with male pattern hair loss to recovery and see if the treatment is effective and safe and how well the drug is tolerated by subjects.
Within this study, the Clascoterone solution will be compared to a placebo.
The study has 2 parts:
Part 1 will see if Clascoterone solution is effective and safe compared to a placebo when applied twice daily for up to 6 months.
Part 2 will see the long-term safety and efficacy of the Clascoterone solution compared to placebo for additional 6 months in subjects defined as ''responders'' in Part 1. A responder is defined as someone who have responded to the study drug, based on research data.
Part 1 of the study is double-blind, meaning that neither the subject nor the study doctor knows which treatment subject is receiving. Part 2 of the study is single-blind and only the study doctor doing the study knows which treatment subject is receiving.
Part 1 of the study will start with baseline visit during which subjects will be randomly assigned (by chance) in ratio 2:1 to apply either Clascoterone or placebo solution to their balding areas of the scalp. Subjects will have 5 clinic visits and 2 follow-up phone calls during 6 months of Part 1 duration. Subjects identified as Part 1 responders at Month 6 visit will be again randomly assigned in ratio 2:1 to receive either study drug or placebo.
Part 2 of the study will consist of 2 additional clinic visits and treatment will last for further 6 months.
Each subject will have also an end of study visit one month after the study drug treatment has been completed or discontinued (it will be one month after end of Part 1 for not responder subjects).
For those subjects who complete the whole study (Part 1 and Part 2), the total duration of the study will be about 14 months, with 12 months of treatment with a total of eight clinic visits and two phone calls.
Subjects taking part in this study will have the medical tests or procedures described below.
* They will be asked about their previous medical history and current medications.
* A brief physical examination will be performed.
* Vital signs, weight and height will be measured.
* Electrocardiograms will be performed.
* Subject's scalp will be checked for any signs of irritation.
* Two different types of photos will be taken during this study: "global photos", i.e. general photos of the subject's scalp and "macro photos", i.e. close up photos of a region of the subject's scalp. Global photos will be taken to help the subject and the study doctor to assess whether there has been a change in subject's hair growth. Macro photos will be used to count the number of hairs in a region of the subject's scalp and measure other properties of the hair (hair width and hair darkness).
* Blood draws and urine sample collection for safety laboratory tests.
* Subject will be asked to complete, on site, the following two questionnaires:
* Cosmetic Evaluation - a couple of cosmetic questions on acceptability and how easy the study drug is to use.
* Male Androgenetic Alopecia Questionnaire - some questions about subject's hair assessment.
Eligible subjects will be given a supply of the study drug and shown how to use and store it. The first study drug dose will be applied at the clinic under the supervision of the study staff. Subjects will be instructed to apply about 1.5 ml of study drug with a dropper to the balding areas of the scalp on the vertex and the temples twice daily, once in the morning and once in the evening.
Subjects will be asked to bring back all used containers of study drug and all unused study drug to each study visit. Subjects will also be given a diary, shown what things have to be recorded on it and asked to bring back the completed diary to the study center at each visit.
Within this study, the Clascoterone solution will be compared to a placebo.
The study has 2 parts:
Part 1 will see if Clascoterone solution is effective and safe compared to a placebo when applied twice daily for up to 6 months.
Part 2 will see the long-term safety and efficacy of the Clascoterone solution compared to placebo for additional 6 months in subjects defined as ''responders'' in Part 1. A responder is defined as someone who have responded to the study drug, based on research data.
Part 1 of the study is double-blind, meaning that neither the subject nor the study doctor knows which treatment subject is receiving. Part 2 of the study is single-blind and only the study doctor doing the study knows which treatment subject is receiving.
Part 1 of the study will start with baseline visit during which subjects will be randomly assigned (by chance) in ratio 2:1 to apply either Clascoterone or placebo solution to their balding areas of the scalp. Subjects will have 5 clinic visits and 2 follow-up phone calls during 6 months of Part 1 duration. Subjects identified as Part 1 responders at Month 6 visit will be again randomly assigned in ratio 2:1 to receive either study drug or placebo.
Part 2 of the study will consist of 2 additional clinic visits and treatment will last for further 6 months.
Each subject will have also an end of study visit one month after the study drug treatment has been completed or discontinued (it will be one month after end of Part 1 for not responder subjects).
For those subjects who complete the whole study (Part 1 and Part 2), the total duration of the study will be about 14 months, with 12 months of treatment with a total of eight clinic visits and two phone calls.
Subjects taking part in this study will have the medical tests or procedures described below.
* They will be asked about their previous medical history and current medications.
* A brief physical examination will be performed.
* Vital signs, weight and height will be measured.
* Electrocardiograms will be performed.
* Subject's scalp will be checked for any signs of irritation.
* Two different types of photos will be taken during this study: "global photos", i.e. general photos of the subject's scalp and "macro photos", i.e. close up photos of a region of the subject's scalp. Global photos will be taken to help the subject and the study doctor to assess whether there has been a change in subject's hair growth. Macro photos will be used to count the number of hairs in a region of the subject's scalp and measure other properties of the hair (hair width and hair darkness).
* Blood draws and urine sample collection for safety laboratory tests.
* Subject will be asked to complete, on site, the following two questionnaires:
* Cosmetic Evaluation - a couple of cosmetic questions on acceptability and how easy the study drug is to use.
* Male Androgenetic Alopecia Questionnaire - some questions about subject's hair assessment.
Eligible subjects will be given a supply of the study drug and shown how to use and store it. The first study drug dose will be applied at the clinic under the supervision of the study staff. Subjects will be instructed to apply about 1.5 ml of study drug with a dropper to the balding areas of the scalp on the vertex and the temples twice daily, once in the morning and once in the evening.
Subjects will be asked to bring back all used containers of study drug and all unused study drug to each study visit. Subjects will also be given a diary, shown what things have to be recorded on it and asked to bring back the completed diary to the study center at each visit.
开始日期2023-08-17 |
申办/合作机构 Cassiopea SpA [+3] |
NCT05910450
A 6-month Phase 3, Multicenter, Prospective, Randomized, Double-Blind, Vehicle-Controlled Study, to Evaluate the Efficacy and Safety of Topically Applied Clascoterone Solution for the Treatment of Androgenetic Alopecia in Males, Followed by a 6-month Single-Blind Treatment With Clascoterone or Vehicle Solution
The purpose of the study is to see if Clascoterone can help people with male pattern hair loss to recovery and see if the treatment is effective and safe and how well the drug is tolerated by subjects.
Within this study, the Clascoterone solution will be compared to a placebo.
The study has 2 parts:
Part 1 will see if Clascoterone solution is effective and safe compared to a placebo when applied twice daily for up to 6 months.
Part 2 will see the long-term safety and efficacy of the Clascoterone solution compared to placebo for additional 6 months in subjects defined as ''responders'' in Part 1. A responder is defined as someone who have responded to the study drug, based on research data.
Part 1 of the study is double-blind, meaning that neither the subject nor the study doctor knows which treatment subject is receiving. Part 2 of the study is single-blind and only the study doctor doing the study knows which treatment subject is receiving.
Part 1 of the study will start with baseline visit during which subjects will be randomly assigned (by chance) in ratio 2:1 to apply either Clascoterone or placebo solution to their balding areas of the scalp. Subjects will have 5 clinic visits and 2 follow-up phone calls during 6 months of Part 1 duration. Subjects identified as Part 1 responders at Month 6 visit will be again randomly assigned in ratio 2:1 to receive either study drug or placebo.
Part 2 of the study will consist of 2 additional clinic visits and treatment will last for further 6 months.
Each subject will have also an end of study visit one month after the study drug treatment has been completed or discontinued (it will be one month after end of Part 1 for not responder subjects).
For those subjects who complete the whole study (Part 1 and Part 2), the total duration of the study will be about 14 months, with 12 months of treatment with a total of eight clinic visits and two phone calls.
Subjects taking part in this study will have the medical tests or procedures described below.
* They will be asked about their previous medical history and current medications.
* A brief physical examination will be performed.
* Vital signs, weight and height will be measured.
* Electrocardiograms will be performed.
* Subject's scalp will be checked for any signs of irritation.
* Two different types of photos will be taken during this study: "global photos", i.e. general photos of the subject's scalp and "macro photos", i.e. close up photos of a region of the subject's scalp. Global photos will be taken to help the subject and the study doctor to assess whether there has been a change in subject's hair growth. Macro photos will be used to count the number of hairs in a region of the subject's scalp and measure other properties of the hair (hair width and hair darkness).
* Blood draws and urine sample collection for safety laboratory tests.
* Subject will be asked to complete, on site, the following two questionnaires:
* Cosmetic Evaluation - a couple of cosmetic questions on acceptability and how easy the study drug is to use.
* Male Androgenetic Alopecia Questionnaire - some questions about subject's hair assessment.
Eligible subjects will be given a supply of the study drug and shown how to use and store it. The first study drug dose will be applied at the clinic under the supervision of the study staff. Subjects will be instructed to apply about 1.5 ml of study drug with a dropper to the balding areas of the scalp on the vertex and the temples twice daily, once in the morning and once in the evening.
Subjects will be asked to bring back all used containers of study drug and all unused study drug to each study visit. Subjects will also be given a diary, shown what things have to be recorded on it and asked to bring back the completed diary to the study center at each visit.
Within this study, the Clascoterone solution will be compared to a placebo.
The study has 2 parts:
Part 1 will see if Clascoterone solution is effective and safe compared to a placebo when applied twice daily for up to 6 months.
Part 2 will see the long-term safety and efficacy of the Clascoterone solution compared to placebo for additional 6 months in subjects defined as ''responders'' in Part 1. A responder is defined as someone who have responded to the study drug, based on research data.
Part 1 of the study is double-blind, meaning that neither the subject nor the study doctor knows which treatment subject is receiving. Part 2 of the study is single-blind and only the study doctor doing the study knows which treatment subject is receiving.
Part 1 of the study will start with baseline visit during which subjects will be randomly assigned (by chance) in ratio 2:1 to apply either Clascoterone or placebo solution to their balding areas of the scalp. Subjects will have 5 clinic visits and 2 follow-up phone calls during 6 months of Part 1 duration. Subjects identified as Part 1 responders at Month 6 visit will be again randomly assigned in ratio 2:1 to receive either study drug or placebo.
Part 2 of the study will consist of 2 additional clinic visits and treatment will last for further 6 months.
Each subject will have also an end of study visit one month after the study drug treatment has been completed or discontinued (it will be one month after end of Part 1 for not responder subjects).
For those subjects who complete the whole study (Part 1 and Part 2), the total duration of the study will be about 14 months, with 12 months of treatment with a total of eight clinic visits and two phone calls.
Subjects taking part in this study will have the medical tests or procedures described below.
* They will be asked about their previous medical history and current medications.
* A brief physical examination will be performed.
* Vital signs, weight and height will be measured.
* Electrocardiograms will be performed.
* Subject's scalp will be checked for any signs of irritation.
* Two different types of photos will be taken during this study: "global photos", i.e. general photos of the subject's scalp and "macro photos", i.e. close up photos of a region of the subject's scalp. Global photos will be taken to help the subject and the study doctor to assess whether there has been a change in subject's hair growth. Macro photos will be used to count the number of hairs in a region of the subject's scalp and measure other properties of the hair (hair width and hair darkness).
* Blood draws and urine sample collection for safety laboratory tests.
* Subject will be asked to complete, on site, the following two questionnaires:
* Cosmetic Evaluation - a couple of cosmetic questions on acceptability and how easy the study drug is to use.
* Male Androgenetic Alopecia Questionnaire - some questions about subject's hair assessment.
Eligible subjects will be given a supply of the study drug and shown how to use and store it. The first study drug dose will be applied at the clinic under the supervision of the study staff. Subjects will be instructed to apply about 1.5 ml of study drug with a dropper to the balding areas of the scalp on the vertex and the temples twice daily, once in the morning and once in the evening.
Subjects will be asked to bring back all used containers of study drug and all unused study drug to each study visit. Subjects will also be given a diary, shown what things have to be recorded on it and asked to bring back the completed diary to the study center at each visit.
开始日期2023-06-21 |
申办/合作机构 Cassiopea SpA [+3] |
NCT03665194
A Randomised, Double Blind, Placebo Controlled Phase I Study to Investigate the Effects of Systemically Absorbed Cortexolone 17α-propionate (and Its Metabolites) on QT Interval Following Repeat Topical Administration in Healthy Volunteers
This is a randomised, double blind placebo controlled Phase I study which will assess the safety, tolerability and PK of Cortexolone 17α-propionate (and its metabolites), and its effects on the QTc interval when administered as multiple doses to healthy adult volunteers. Volunteers will receive a morning and evening dose (12 hours apart) of 225 mg (3 mL Cortexolone 17α-propionate applied topically as a 7.5 % solution (75 mg in 1 mL), giving a total daily dose of 450 mg (6 mL) per individual.
开始日期2018-06-06 |
申办/合作机构 |
100 项与 Cassiopea SpA 相关的临床结果
登录后查看更多信息
0 项与 Cassiopea SpA 相关的专利(医药)
登录后查看更多信息
10
项与 Cassiopea SpA 相关的新闻(医药)2025-05-30
作者|momo2025年5月,三生制药以一场震撼行业的“王炸级”操作赢麻了,60.5亿美元天价BD交易刷新国产创新药出海天花板,其PD-1/VEGF双抗SSGJ-707凭借“Best-in-Class”临床数据成为“下一个K药”有力竞争者。 01 天价BD,孕育下一个K药2025年5月20日,中国生物医药行业迎来历史性时刻——三生制药宣布与辉瑞达成总额60.5亿美元的授权协议,将其PD-1/VEGF双抗SSGJ-707的全球(除中国内地)权益授予辉瑞。这一交易以12.5亿美元首付款刷新国产创新药出海纪录,远超此前康方生物AK112与Summit Therapeutics达成的50亿美元总交易规模(图1)。三生制药SSGJ-707正在PD-1赛道上演与K药的“王座继承战”。图1.三生制药与辉瑞达成总额60.5亿美元的授权协议SSGJ-707的成功源于其独特的技术设计与临床数据优势。作为三生制药基于专有CLF2平台开发的双抗药物,其采用天然IgG4结构设计,通过消除ADCC和CDC效应,显著降低了免疫系统误伤正常组织的风险,从而在安全性上优于同类竞品。这一差异化设计使其在临床中展现出“Best-in-Class”潜力。图2.SSGJ-707 临床早期数据2025年JPM大会披露的II期数据显示,在单药一线治疗PD-L1阳性非小细胞肺癌(NSCLC)患者中,SSGJ-707的客观缓解率(ORR)达70.8%,疾病控制率(DCR)为100%;联合化疗时,鳞癌患者ORR更飙升至81.3%,且3级及以上不良反应发生率仅为23.5%(图2)。相比之下,康方生物的AK112在III期临床试验中ORR为50%,安全性数据亦稍逊(3级及以上不良反应发生率29.4%)。尽管两者尚未开展头对头试验,但SSGJ-707的疗效与安全性组合已为其赢得“BIC”标签。图3.PD-(L)1/VEGF双抗全球研发进展PD-1/VEGF双抗的崛起源于其能够同时抑制免疫检查点PD-1和促血管生成因子VEGF“双重抗癌机制”。康方生物AK112于2024年9月公布的HARMONi-2 III期研究数据首次在头对头试验中击败默沙东的K药,成为该赛道的引爆点。此后,全球药企加速布局,而三生制药凭借SSGJ-707的快速推进(全球进度第二)抢占先机。目前,全球在研PD-(L)1/VEGF双抗达35款,其中中国占据20款(图3),但仅康方AK112上市,三生(SSGJ-707)、礼新(LM-299)、普米斯(PM8002)等紧随其后。辉瑞选择SSGJ-707而非进度更早的AK112,既因其临床数据亮眼,亦与其适应症拓展潜力及联合疗法开发空间密切相关。 02 展开合作,扩大治疗领域近年来,三生制药通过密集的对外合作与产品引进,持续拓宽治疗领域边界,逐步构建起覆盖多疾病领域的创新产品管线(图4)。在肿瘤领域,三生制药通过ADC、双抗等前沿技术组合探索治疗潜力。2025年2月,三生制药与百利天恒达成合作,共同推进其PD-1/VEGF双抗SSGJ-707与百利天恒全球首创的EGFR×HER3双抗ADC药物BL-B01D1的联用研究。BL-B01D1作为全球唯一进入临床阶段的靶向EGFR×HER3双抗ADC,已在中美开展约30项临床试验,覆盖非小细胞肺癌、乳腺癌等适应症,其中联合奥希替尼用于一线非小细胞肺癌的III期试验已完成首例受试者入组。此外,2025年1月,三生制药引进映恩生物HER2 ADC药物DB-1303,获得其在中国内地及港澳的商业化权利。DB-1303采用拓扑异构酶-1抑制剂载荷,已获FDA及NMPA突破性疗法认定,针对HER2低表达乳腺癌的III期临床试验全球领先,疾病控制率(DCR)高达94.1%。图4.公司近年来开展的合作项目(部分)在血液疾病领域,三生制药聚焦中国高发的急性髓性白血病(AML)。2024年11月,三生制药获得东阳光药第二代FLT3抑制剂苯磺酸克立福替尼的独家商业化权利。该药物针对FLT3-ITD突变型AML,其III期临床试验数据显示更强的靶点抑制活性及更低脱靶风险,有望成为国内首个上市的国产高选择性FLT3抑制剂。 2024年5月,三生蔓迪与翰宇药业合作开发司美格鲁肽注射液,切入千亿减重市场(图5)。该产品是国内首个进入III期临床的国产司美格鲁肽类似物,依托三生制药的营销网络,有望在超重/肥胖率超50%的中国市场快速渗透。同时,公司在痛风领域从EnzymeRx 引进的SEL-212(Pegsiticase)已于2024年7月向FDA提交生物制品许可申请(BLA),其通过重组酶代谢尿酸的机制为顽固性痛风患者提供新选择,潜在市场空间达百亿级。三生制药在剂型革新上亦取得显著进展。2024年10月,三生获得海和药物紫杉醇口服溶液(柏瑞素)的独家商业化权利(图5)。该产品采用脂质自乳化技术(DH-LASED),突破紫杉醇传统注射限制,生物利用度提升且过敏风险降低,无需预处理即可居家服用,极大提升晚期胃癌患者依从性。其针对HER2阴性乳腺癌的国际多中心III期研究已进入随访阶段,未来适应症拓展潜力显著。 图5.三生制药在多个领域展开密集合作在血小板减少症领域,三生制药与则正医药合作的艾曲泊帕乙醇胺干混悬剂(特艾升)于2024年12月获批上市。该干混悬剂型适口性更佳,支持精准剂量调整,尤其适合儿童及吞咽困难患者,进一步巩固了公司在骨髓保护产品线的领导地位(图5)。更令人关注的是,三生制药通过引进Cosmo 附属公司 Cassiopea 旗下Winlevi(柯拉特龙乳膏)进一步切入痤疮治疗领域。作为40年来首个新机制外用雄激素受体抑制剂,Winlevi在美国市场处方量已超109万张,其在中国内地的III期桥接试验预计2025年完成,有望成为国内首个AR拮抗剂类痤疮药物,覆盖中重度患者群体。 03 发力自免,创造第二增长曲线随着全球自身药物市场加速扩容,三生制药控股子公司三生国健作为国内自免领域先行者,已构建覆盖IL-17A、IL-4R、IL-5、IL-1β等核心靶点的产品矩阵,并前瞻性布局BDCA2、TL1A等下一代靶点,推动自免业务成为继肿瘤领域后的第二增长曲线。其中,SSGJ-608是是国内进度第二的IL-17A单抗,针对斑块状银屑病,为全新的氨基酸序列,在体外和体内动物模型中显示出和同靶点抗体Cosentyx 和 Taltz 相当的生物活性。其针对中重度斑块状银屑病的III期临床试验显示,12周主要疗效数据优异,短期内快速起效、疗效优势明显、维持治疗期,SSGJ-608给药间隔延长至Q4W或Q8W疗效持续维持高位,有望在PsO上实现更长给药间隔(图6)。图6.SSGJ-608 III期临床研究数据相较于已上市的司库奇尤单抗(诺华)和依奇珠单抗(礼来),SSGJ-608通过优化氨基酸序列实现更高生物活性,并计划拓展中轴型脊柱炎适应症,预计2025年递交NDA,成为国产IL-17A领域的重要竞争者。此外,SSGJ-611是全球第三款靶向IL-4Rα的生物制剂。它通过精准阻断IL-4/IL-13炎症信号通路,抑制2型免疫反应过度激活,从而快速缓解瘙痒、修复皮肤屏障。在特应性皮炎(AD)II期临床中(图7),300mg每两周给药组的EASI-75应答率达72.5%,优于度普利尤单抗同阶段数据(67%),并计划于2026年提交NDA。其适应症布局覆盖AD、COPD及慢性鼻窦炎伴鼻息肉(CRSwNP),目标患者群体超1.6亿人,有望复制度普利尤单抗“一药多适应症”的放量路径。图7.SSGJ-611 II期临床研究数据值得注意的是,针对急性痛风性关节炎的重组抗白介素1-β(IL-1β)人源化单克隆抗体SSGJ-613的II期临床结果显示,使用SSGJ-613治疗12周复发率仅8.3%,显著低于阳性对照组倍他米松(28.6%),且疼痛评分降幅达70%以上。目前国内尚无IL-1β单抗上市,长春高新的金纳单抗虽已递交NDA,但SSGJ-613凭借更优疗效数据和三生国健在风湿科的渠道优势,有望在2025年NDA后快速抢占市场。与此同时,三生国建持续布局BDCA2、TL1A等自免领域前沿靶点。BDCA2靶点通过抑制浆细胞样树突状细胞(pDC)产生干扰素α/β,直击红斑狼疮核心病理机制。三生国健的SSGJ-626已在中美同步获批IND,进度仅次于Biogen的Litifilimab(III期临床中),有望成为全球第二款进入临床的BDCA2单抗(图8)。图8.SSGJ-626有望成为全球第二款进入临床的BDCA2单抗另外,TL1A靶点在溃疡性结肠炎、克罗恩病等适应症中展现突破性潜力,辉瑞、默沙东等跨国药企已通过BD交易押注该赛道。三生制药的SSGJ-627于2025年1月获准IND,成为国内首个进入临床的TL1A单抗,有望在消化系统自免领域建立先发优势。 结语 三生制药凭60.5亿美元天价BD交易、SSGJ-707强劲数据及肿瘤、自免多线布局实力“赢麻”,以创新硬实力与前瞻战略,在出海与本土市场双制霸,成国产药企“顶流”标杆!参考资料[1]https://www.3sbio.com/ImgUpload/files/202501/2025012104430567755.pdf[2]https://www.3sbio.com/ImgUpload/files/202408/2024082206294824405.pdf[3]三生制药官网、官微、国盛证券研报、中泰证券研报、各种公开资料等共建Biomedical创新生态圈!如何加入BiG会员?
2025-05-06
◆ ◆ ◆ ◆Paul's Insight2025年4月28日-5月4日◆ ◆ ◆ ◆过去一周(4月28日至5月4日),全球制药监管与行业机构发布了多项重要动态。美国FDA在细胞产品安全和质量管理等领域持续发力;欧盟EMA在临床前新方法学新药审批上有新的更新;欧洲药典EDQM进一步完善CEP电子提交流程;英国MHRA在疫苗审批、给药创新和执法行动上齐头并进;美国PDA重启区域分会,并探讨前沿疫苗传递技术;ISPE则描绘了生物制造设施与数字化转型的未来蓝图。美国FDA细胞产品安全和质量/GMP两大领域持续发力1. 人源细胞组织产品安全管理新指南草案5月2日,美国FDA的生物制品评估与研究中心(CBER)发布两份针对人源细胞、组织及衍生产品(HCT/P)的指南草案。一是《减少HCT/P相关脓毒症病原体传播风险的建议》,更新了2007年供体资格决策中脓毒症筛查与检测标准;二是《减少HCT/P中结核分枝杆菌传播风险的建议》,在专用检测工具面世前,明确了结核病史与风险因素评估的推荐措施。两份草案现正面向公众征询意见,反馈截止日期为7月7日。图:美国FDA发布两份针对人源细胞、组织及衍生产品(HCT/P)的指南草案https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-reduce-risk-transmission-disease-agents-associated-sepsis-human-cells-tissues-and2. 质量管理成熟度原型评估计划FDA药品评估与研究中心(CDER)宣布,将启动自愿质量管理成熟度(QMM)原型评估计划的第二轮工作。该计划面向符合条件的药品生产企业开放申请,申请者需具备已完成注册、具备检查记录,并在美国市场有商业化分销产品等资格。入选企业将接受由三名CDER工作人员组成的QMM评估团队的系统性评估,评估不涉及负责cGMP合规的FDA检查人员。为期五天的评估将围绕企业的质量管理实践进行,企业将提前收到评估议程,并需安排适当人员参与。评估结束后,企业将收到一份详尽的评估报告,内容包括各实践领域的评分、说明、优势亮点及改进建议,并有机会与评估团队面对面沟通报告内容、提出问题或反馈意见。图:FDA启动质量管理成熟度原型评估计划第二轮工作https://www.federalregister.gov/documents/2025/04/23/2025-06968/voluntary-quality-management-maturity-prototype-assessment-protocol-evaluation-program3. 电商平台执法力度再升级4月29日,FDA向电商巨头亚马逊发出警告信,指控其平台销售未经FDA批准的新药,包括多款声称用于纹身前后镇痛的外用制剂。FDA通过亚马逊网站采购样品后确认,相关产品均未获FDA许可且标签不合规,构成对《联邦食品、药品和化妆品法案》第301条的违反。此次行动彰显FDA对线上销售渠道监管的持续加码。图:FDA向电商巨头亚马逊发出警告信https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/amazoncom-inc-689355-041720254. 泛疫苗(广谱疫苗)新体系——“金标准”项目5月1日,美国卫生部(HHS)和国立卫生研究院(NIH)联合宣布启动“金标准”(Generation Gold Standard)泛疫苗开发项目,计划投入约5亿美元,研发可同时对抗多种流感和冠状病毒株的广谱疫苗。项目将采用β-丙内酯灭活全病毒技术,目前已在实验室中制备了针对H5N1与多种冠状病毒的候选疫苗。 图:美国官方宣布开发泛疫苗(广谱疫苗)新体系https://www.hhs.gov/press-room/hhs-nih-announces-generation-gold-standard.html美国PDA分会动态与疫苗给药创新1. 波多黎各分会重新启动5月2日,PDA发表文章宣布重启波多黎各分会,新任负责人Myrta Atiles将聚焦AI等新兴技术应用,推动区域人才培养与技术创新。2月25日于波多黎各举办的“加速创新,快速惠及患者”会议,探讨了无菌加工、技术转移敏捷性及监管加速等话题。波多黎各是全球重要的制药制造中心之一,拥有高度集中的制药和生物制药产业,对该地区经济具有支柱性作用。图:PDA宣布波多黎各分会重新启动https://www.pda.org/pda-letter-portal/home/full-article/pda-puerto-rico-chapter-reignites2. 微针阵列贴片(MAP)疫苗给药创新4月29日,PDA发布文章聚焦微针阵列贴片(MAP)这一新兴疫苗传递技术。MAP通过不足一毫米的微针将疫苗递送至皮肤表层,激活免疫反应,具备减少剂量、简化冷链物流、提高接种率等优势。其材料多为可溶性或可降解聚合物,具备良好生物相容性。临床研究显示,MAP在安全性和免疫效果上表现优异,尤其适用于资源有限地区。尽管仍面临生产、监管等挑战,MAP有望成为未来全球疫苗接种的重要工具。图:PDA探讨疫苗给药创新https://www.pda.org/pda-letter-portal/home/full-article/microneedle-array-patches-for-vaccine-delivery欧盟EMA临床前研究新方法学与新药审批1. 推动新方法学(NAM)替代动物试验欧洲药品管理局(EMA)表示,其正积极推动在药物研发和测试中采用新的方法学(NAM),以减少动物使用,践行“3R”原则(替代、减少、优化)。这些新技术包括器官芯片、类器官、体外细胞模型和计算机建模,旨在在确保药物安全有效的同时,提高科学预测性并减少伦理争议。EMA通过其创新工作组(ITF)为开发者提供免费咨询平台,帮助新方法更顺利地被纳入监管体系。图:EMA推动新方法学替代动物试验https://www.ema.europa.eu/en/human-regulatory-overview/research-development/ethical-use-animals-medicine-testing/regulatory-acceptance-new-approach-methodologies-nams-reduce-animal-use-testing2. 对Winlevi(clascoterone)给出负面意见4月25日,EMA人用药品委员会(CHMP)对意大利Cassiopea公司申请的痤疮治疗药Winlevi(clascoterone)提出负面意见,认为其在12至18岁青少年中的潜在风险大于获益。Winlevi作为一种新型雄激素受体阻断药,虽以乳膏形式用于治疗寻常痤疮,但可能抑制下丘脑、垂体及肾上腺功能,影响青少年的生长发育与性成熟。尽管企业提交的数据显示风险较低,并提出相应的风险控制措施,CHMP仍认为证据不足,最终建议拒绝其上市许可申请。图:EMA对痤疮治疗药的评审提出负面意见https://www.ema.europa.eu/en/medicines/human/EPAR/winlevi欧洲EDQMCEP电子化提交流程再升级5月2日,EDQM宣布,将接受基于eCTD验证标准v8.1的CEP申请,同时在2025年5月31日前继续兼容v7.1版本,以确保提交过程平稳过渡。此举旨在应对当前EU M1包中跟踪表位置要求与EDQM实际流程不一致的问题。图:CEP电子化提交流程再升级https://www.edqm.eu/en/w/validation-of-ectd-submissions-for-cep-applications英国MHRA新药审评与打击非法药品行动1. Vimkunya基孔肯雅疫苗获批5月1日,英国药品和健康产品监管局(MHRA)通过国际认可程序(IRP)批准Vimkunya疫苗,用于预防12岁及以上人群感染基孔肯雅病毒。基孔肯雅病由受感染蚊子传播,常见于亚洲、非洲及美洲的亚热带地区,患者通常出现发热、皮疹及关节剧烈疼痛,症状可能持续数月甚至数年。Vimkunya疫苗的批准基于IRP机制,该机制允许MHRA借鉴国际可信监管机构的专业评估,以加快英国患者的用药可及性。MHRA在IRP框架下仍保留对证据强度不足的申请予以拒绝的权力。图:MHRA通过国际认可程序(IRP)批准疫苗https://www.gov.uk/government/news/vimkunya-vaccine-approved-to-prevent-disease-caused-by-the-chikungunya-virus-in-people-12-years-of-age-and-older2. 纳武利尤单抗皮下注射新给药方案批准4月30日,英国MHRA批准纳武利尤单抗(nivolumab,商品名Opdivo)皮下注射新配方,将给药时间从传统的30–60分钟静脉输注大幅缩短至仅需3–5分钟,适用于包括肺癌、黑色素瘤等多种肿瘤类型,显著提升患者治疗舒适度与临床操作效率。该药由百时美施贵宝(BMS)研发,是一款程序性死亡受体-1(PD-1)抑制剂,可通过激活免疫系统识别并攻击肿瘤细胞。图:纳武利尤单抗皮下注射给药方案批准https://www.gov.uk/government/news/mhra-authorises-cancer-treatment-variation-with-an-administration-time-of-3-5-minutes3. 打击非法药品4月29日凌晨,英国MHRA在英格兰中西部及西北部发起大规模突袭行动,逮捕了12名涉嫌组织药品贩运的犯罪嫌疑人。这也是MHRA历史上规模最大的一次此类刑事调查。执法人员在现场查获了大量非法药物和犯罪资产:数十万剂包括类鸦片镇痛药和抗焦虑药在内的受控药物被缴获。图:英国MHRA打击非法药品行动https://www.gov.uk/government/news/twelve-arrested-in-mhras-biggest-ever-crackdown-on-organised-medicines-trafficking国际ISPE上周,ISPE发布了两篇预告文章,介绍2025年ISPE生物技术大会将重点讨论的两大主题:1. 生物制造设施的未来格局在“设施生命周期”专场中,行业专家将探讨如何从传统的静态基础设施,转型为灵活、自适应的生产生态系统,以应对多样化治疗模式对速度、可持续性和敏捷制造的需求。重点议题包括:武田制药展示的实时产能建模、模块化建设与数字化集成实践,以及如何通过应用ISO 14644-16等国际标准,实现洁净室的节能设计和运营效率最大化。2. 人工智能/机器学习与生物技术融合趋势在“利用人工智能和机器学习(AI/ML) 加速生物制药发展”专场,专家们将分享AI/ML在药物研发、临床试验优化、智能制造及供应链管理中的应用实例,展示其如何加速产品上市并提升运营效率。讨论内容包括支持AI部署的数据架构建设、AI驱动的持续工艺确认(CPV)与实时放行(RTR)、mRNA项目中的可解释性建模,以及通过强化学习实现自主运营与技能传承的前沿探索。图:ISPE发布了预告文章,介绍2025年ISPE生物技术大会将重点讨论的主题https://ispe.org/pharmaceutical-engineering/ispeak/trends-and-opportunities-how-artificial-intelligence-ai-and结 语回顾一周动态,各大监管机构和行业组织在药品安全、监管创新、技术应用和执法合规等方面均有进展。FDA聚焦细胞治疗与质量管理、EMA强调替代动物试验与风险评估、EDQM优化电子提交流程、MHRA加速疫苗与给药方案审批并加强执法、PDA与ISPE则在疫苗传递与生物制造数字化等方面绘制蓝图。这些举措体现了全球制药监管与行业在科学进步、伦理责任和患者需求三者之间的协调与平衡,共同推动药品研发和监管迈向更加智能、高效和安全的未来。END声明:本文仅代表作者个人观点,不代表任何组织及本公众号立场,如有不当之处,敬请指正。如需转载,请注明作者及来源:蒲公英Biopharma。活动推荐5月8日 | 线上 | 双维度解锁无菌药品生产污染防控策略5月16日 | 线上 |《VHP生物指示剂的应用》5月22日 | 线下 | 2025中国无菌药品制造创新发展论坛
疫苗基因疗法加速审批
2023-04-01
·动脉网
2023年以来,两家中国公司通过发行GDR在瑞士上市,成为瑞士证券交易所(以下简称“瑞交所”)2023年到目前为止仅有的两家公司上市。而2022年至今,已有11家中国公司发行GDR并在瑞交所上市,募资总额35.5亿美元(约240多亿元人民币),另有40多家公司正在筹备或已着手申请。赴瑞士上市的热潮始于一项新政,2022年,中国证监会发布《境内外证券交易所互联互通存托凭证业务监管规定》,将“沪伦通机制”扩展至“中欧通机制”,从政策层面给予中国企业出海融资大力支持。这股热潮自然也席卷了医疗行业。2022年9月,乐普医疗、健康元在瑞交所上市;截至目前,爱博医疗、鱼跃医疗、康希诺、百克生物、奕瑞科技相继筹划或推进瑞士上市事宜。一年时间内吸引数十家中国企业前往,瑞交所究竟有何魅力?对于医疗行业来说,企业选择瑞交所上市的意义何在?且看本文分析。 国际化战略落地,境外上市成为重要途径企业在境外上市融资,与其业务布局密不可分。目前,已在瑞士上市和拟上市的几家医疗健康企业,都已不同程度地涉足了海外业务。 7家公司的海外业务布局,资料来源:公司财报(部分公司尚未发布2022年财报,以2021年财报信息为例)这些企业中,乐普医疗、健康元、鱼跃医疗等公司已上市产品众多,且在海外进行了广泛的业务布局。乐普医疗2019年成立国际事业部,全面整合资源,拓展海外市场;通过产品的全球化布局,有效降低国内集采政策影响。截至2021年底,乐普医疗累计申请专利1385项,产品获得美国FDA认证33项、欧盟CE认证171项。在持续的国际化战略推动下,2019-2021年,乐普医疗海外业务收入分别为5.54亿、15.42亿和37.6亿元,占比从7.11%提升至35.27%。新冠疫情期间,乐普医疗快速推进了新冠抗原检测产品在CE的准入和销售,带动了抗原检测产品和其他产品的海外销售。健康元的原料药及中间体向亚洲、欧洲、北美、非洲等近40个国家和地区出口;制剂方面,加强了呼吸、辅助生殖、抗真菌、抗病毒、消化道等产品在巴基斯坦、印度尼西亚、菲律宾等国家和地区的准入和推广。鱼跃医疗也设立了海外事业部,强化海外属地化团队建设,外销渠道持续拓宽。此外,奕瑞科技在美国、韩国、日本、德国等地设立了境外分支机构,并在多个国家建立了海外客户服务平台或销售团队。2022年,奕瑞科技实现境外收入5.25亿元,占总营收的33.9%。值得一提的是,奕瑞科技对CMOS传感器、芯片与碘化铯等部分关键原材料的采购相对集中,境外采购占比超过了20%。爱博医疗的国际化战略也在快速推进中,人工晶状体产品上市以来,已出口至德国、法国、荷兰、意大利、奥地利、卢森堡、泰国、巴基斯坦等国家。几家公司中,康希诺的境外收入占比一度达到71.1%。疫情期间,康希诺研发的重组新型冠状病毒疫苗(5型腺病毒载体)在境内和境外多个国家获得紧急使用授权/附条件上市,对营业收入产生积极影响。2021年,康希诺首次实现盈利,其中境外收入30.57亿元,占营业收入的71.1%,主要是向墨西哥、巴基斯坦等国家供应新冠疫苗带来的收入。不过,这仅是疫情期间的特殊情况。2022年,全球新冠疫苗市场环境发生变化,接种增速放缓,部分地区甚至供过于求,康希诺境外收入占比降至21.1%;由于研发费用高达7.78亿元,占总营收的76.35%,康希诺也再次回归亏损状态。对各家企业而言,国际化都已成为长期战略,选择境外上市是实施战略的重要途径。只是对于不同企业而言,境外上市的短期诉求有所不同。乐普医疗、鱼跃医疗等已处于商业化阶段的公司,需扩大在海外的生产、销售和无网络;对于康希诺这样仍保持着高水平研发投入的公司来说,需上市融资以支持后续研发工作,尤其是进一步拓展海外业务、临床试验选择及商务合作机会。生命科学公司集中的瑞交所,有多香?那么,为什么是瑞交所?一个大的背景是:生命科学公司是瑞交所的一大特色。瑞交所母公司SIX集团的首席执行官迪塞尔霍夫(Jos Dijsselhof)曾介绍,瑞交所是欧洲医疗保健公司的主要上市地,制药、生物技术、医疗器械等相关企业约占欧洲生命科学市场的三分之一。几大证券交易所生命科学公司市值情况(单位:10亿欧元)资料来源:SIX and STOXX,截至2020年1月生命科学公司在瑞交所如此集中并非巧合,长期以来,瑞士的制药、生物技术和医疗器械公司都与当地金融机构之间保持良好互动,形成了良性行业生态,并受到国际关注。一批又一批生命科学公司在瑞交所成长起来,瑞交所也吸引着一批又一批颇具影响力的公司前来。全球制药巨头诺华、罗氏在瑞交所上市,为形成多元化的行业生态系统奠定了基础。2000年,瑞士制药公司Actelion在瑞交所上市;2017年,强生斥300亿美元巨资收购了Actelion,并将Actelion的药物发现和早期研发部门分拆出新公司Idorsia,作为对Actelion前股东的实物贡献。Idorsia也于2017年6月在瑞交所上市。瑞交所对跨境上市也极具吸引力,近年来,Newron、Cosmo、IGEA Pharma、Cassiopea等公司在瑞交所上市。同时,瑞士拥有丰富的生命科学集群和强大的生命科学生态体系,其中包括大型制药公司、中小企业以及初创企业、世界领先的大学、行业协会等等。据瑞士贸易与投资处发布的信息显示,瑞士的生命科学领域包括生物技术、制药产业和医疗技术等细分领域。其中,生物技术和制药产业涵盖了整个价值链,并拥有显著增长的生产能力,两大细分领域贡献了瑞士40%以上的出口,使得该产业成为瑞士国民经济的重要支柱。2019年,在瑞士生命科学产业工作的人员达9.5万人,特别是在巴塞尔、莱芒湖地区和苏黎世-楚格地区。因此,中国企业选择瑞士上市,能够依托瑞交所乃至瑞士的生命科学行业生态,助力自身国际化战略加速实施。一些更直接的原因来自上市流程与速度。国内券商分析指出,选择在瑞交所发行GDR与在其他主要市场上市一样,对于上市公司的内控质量报告、审计报告、财务数据报告、管理交易披露等都有相关的披露要求,主要区别点在于:GDR并不被视作金融市场主体公司,相对主体公司进行交易所备案发行,所需材料更少;瑞交所审批时间最快只要20个工作日,GDR项目从启动到完成最快只需要三到四个月左右。中国企业发行GDR上市在瑞士上市流程图片来源:瑞交所官网近年来,受国际大环境影响,赴美上市企业陆续退市撤资,而欧洲市场表现相对平稳,综合产业资源、上市成本等因素的考虑,瑞交所成为了包括医疗行业在内的众多行业A股公司境外上市热门选项。 瑞交所上市,既有资金、也有资源2022年9月,乐普医疗发行的GDR募集资金总额为2.24亿美元,扣除承销费用后实际到账金额为2.20亿美元;健康元发行的GDR募集资金总额约为9204万美元。真金白银能够直接用于海外业务拓展,随之而来的,还有更丰富的海外产业资源、人力资源,以及更强的海外影响力。总的来说,发行GDR并在瑞士上市,对不同企业的海外业务发展都将有特定价值。其一,完善全球研、产、销体系。自主研发具有领先性、竞争力的产品,是创新企业永恒的主题。开拓境外融资渠道,将募集资金用于全球研发,也是企业共同的诉求。获得境外融资之后,企业可更便捷地建设境外研发中心,推进海外临床试验和产品注册。同时,通过授权或共同开发等模式,引入全球领先技术的产品,或将公司重要产品拓展到全球,最大化提升公司产品和研发管线的价值。通过海外生产基地和销售网络的配套建设,企业能降低产品“出海”成本,并提供更及时的客户服务。例如,乐普医疗在瑞士发行GDR上市后,将建设境外产业化生产基地、提高海外产能,设立境外商务拓展中心,建设全球产品销售网络与售后服务体系,以缩短生产半径、降低生产成本、更好地进入属地国家及全球市场。其二,寻求更广阔的并购机会。上市公司依靠并购进行业务扩展、抢占更多市场份额、抬升技术壁垒已是常态,这在以技术创新为核心的器械领域尤其明显。以奕瑞科技所在的数字化 X 线探测器领域为例,海外巨头通过横向并购的方式强强联合,整合优势资源,以巩固竞争力。佳能收购TOSHIBA 医疗(包括旗下探测器业务)、万睿视收购传统巨头珀金埃尔默(Perkin Elmer)影像部件业务等,均是如此。通过投资并购来加强市场地位,也是奕瑞科技的重要规划。奕瑞科技在2022年报中提到,在竞争日益激烈的产业形势下,单纯依靠公司内生发展已经不能满足未来的市场竞争需要。公司考虑在有机成长的同时,通过投资并购与公司业务及未来发展具备战略整合前瞻性的数字X线影像核心部件相关业务,整合全球范围内的前沿技术,进一步建立、完善全球研发、销售、供应链一体化平台,使公司能够覆盖更多的产品品类、占领更多细分市场。综合类器械公司也通过买买买来扩张业务版图、提升技术实力。鱼跃医疗就曾通过收购德国曼吉士(Metrax)切入急救赛道,曼吉士拥有知名的AED品牌普美康(Primedic)。境外上市融资,企业可寻找与自身主业更加契合的全球领先技术,寻求更广阔的并购和投资机会,尤其是在瑞交所这样生命科学公司集中的地方,瑞士本身也是先进医疗器械企业集中的国家。其三,提升品牌海外知名度和影响力。品牌知名度和影响力对企业而言,有着重要的附加价值。提升全球知名度和影响力,对国内企业而言,能在无形中助推全球业务布局、吸引全球客户和人才。而瑞交所在生命科学领域的影响力,将直接带动上市公司的影响力。业内流传着这样的说法:如果说纳斯达克是互联网公司的试金石,那么瑞交所就是医疗的“鉴定书”。以疫苗为例,尽管新冠疫苗热度已“翻篇”,但新冠大流行导致全球各地从政府到大众,更加重视免疫接种,针对尚未被满足的免疫需求,疫苗创新研发仍有着巨大市场空间。尤其是,世卫组织的目标之一是改善全球公共卫生状况,疫苗属于其中的重要部分。而世卫组织总部就设在瑞士,康希诺、百克生物等疫苗企业若在瑞交所成功上市,其影响力将大大提升。2022年,康希诺已经在瑞士设立了欧洲运营中心,从事商务服务业、推广宣传、联盟管理、全球医药事务运营以及市场分析等工作。得益于海外研产销体系完善、影响力提升,企业在全球范围内吸引到更优秀的人才;在海外加强与相应领域的专家合作,设立分支机构、海外人才基地,培养当地的核心技术人员,与目标业务区域的市场、文化结合更紧密,使企业的国际化战略走得更好、更稳。此外,发行GDR和瑞士上市,还能为企业进一步引入境外专业投资机构、产业投资者,优化公司股权结构,持续提升治理透明度和规范化水平。 融资渠道多元化,全球创新步伐加速过去很长时间以来,中国市场以庞大的人口规模、市场空间,成为大型跨国企业的必争之地。在此过程中,受政策导向、市场需求、市场竞争等因素驱动,本土企业创新意识、创新能力提升,也不再满足于国内市场,将视野拓宽到海外。国内市场以进集采、进医保等策略保证市场占有率,海外市场也逐渐从欧美日韩等发达国家逐步扩大至新兴国家。因此,现阶段谈创新,其界定范围已经发生变化:由以往的国内创新变为全球创新,是否具有全球市场内的颠覆性平台、差异化产品或领先技术,成为企业是否具有全球竞争力的关键。而全球创新、维持全球创新能力,又需要充足的资金和资源支持。2022年以来,境内外证券市场新政频出,为企业进行二级市场融资创造了更优越的环境。2022年2月,“沪伦通机制”扩展到“中欧通机制”,将全球存托凭证(GDR)发行地从英国市场拓展至瑞士、德国市场,进一步提升全球资本市场的互联互通水平,彰显资本市场开放信心,让更多优质中国企业拓展境外融资渠道。2023年2月,注册制经过在科创板、创业板、北交所的试点,稳步推向A股全市场,发行上市全过程更加规范、透明、可预期。2023年3月,港交所宣布推出特专科技公司上市机制,降低了已商业化公司和未商业化公司最低市值要求,以及对未商业化公司的研发开支比例的门槛。各大证券交易所对企业所在的细分领域、收入和利润、研发投入等有不同要求,各自优势也不尽相同,整体上为企业提供了多元化的融资渠道。在更加强有力的资金和资源支持下,中国医疗企业的全球创新,定会快马加鞭。参考资料:The Swiss StockExchange:Enjoy the Benefits of Europe’s Leading Exchange for LifeSciences Companies瑞士商务参赞:中瑞是创新型国家 携手生物医药合作华宝证券:海外融资新思路:国内企业出海为何选择 GDR?*封面图片来源:123rf近期推荐声明:动脉网所刊载内容之知识产权为动脉网及相关权利人专属所有或持有。未经许可,禁止进行转载、摘编、复制及建立镜像等任何使用。动脉网,未来医疗服务平台
财报医药出海带量采购
100 项与 Cassiopea SpA 相关的药物交易
登录后查看更多信息
100 项与 Cassiopea SpA 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年11月10日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
申请上市
1
1
其他
登录后查看更多信息
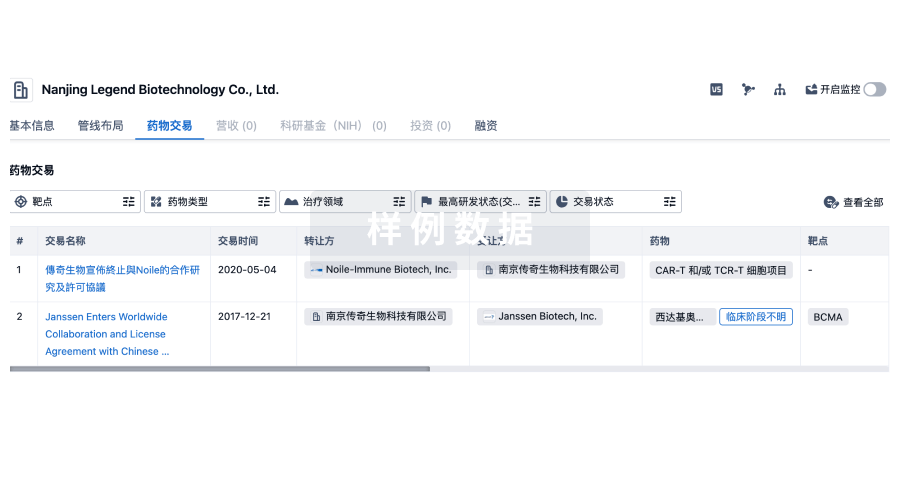
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
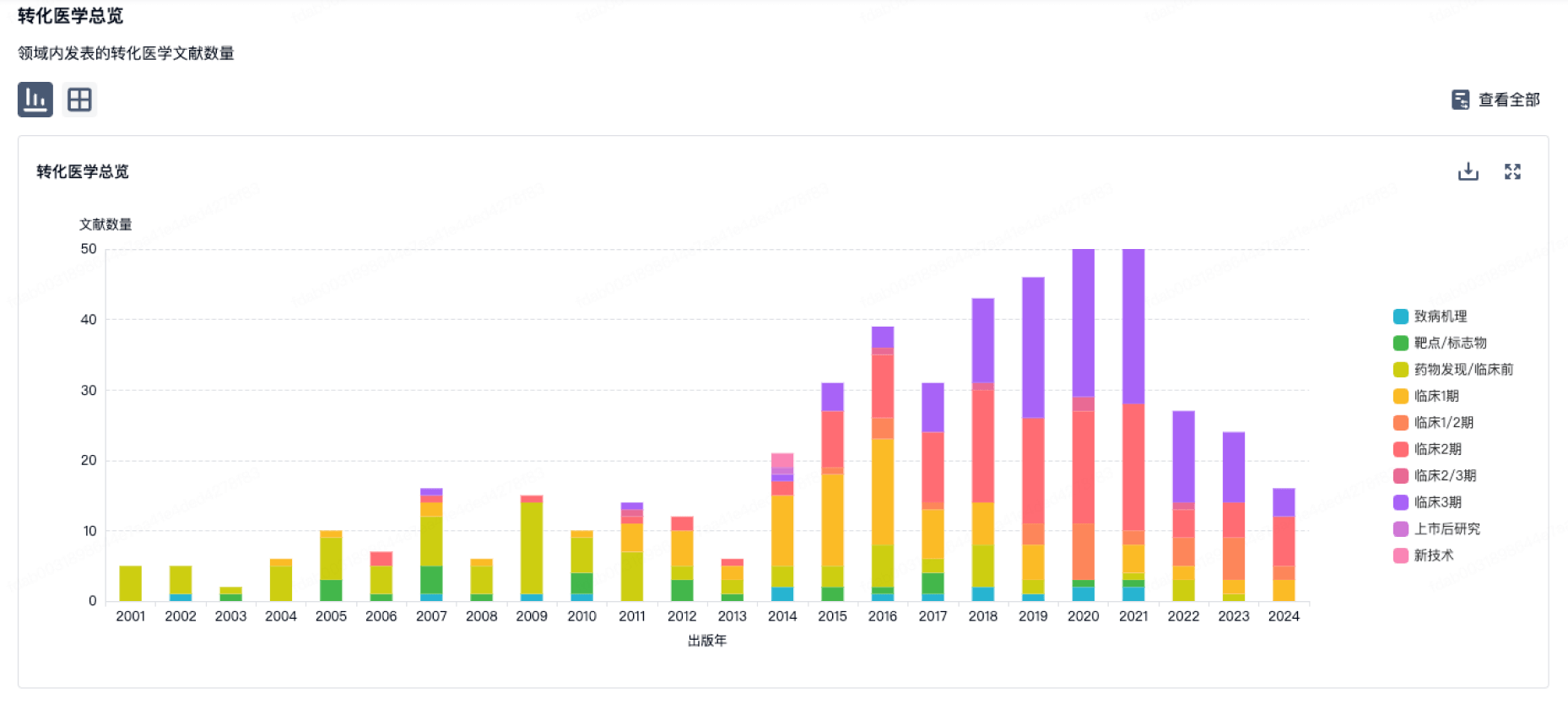
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用