|
|
|
|
|
非在研适应症- |
最高研发阶段临床3期 |
首次获批国家/地区- |
首次获批日期- |
|
|
|
|
|
非在研适应症- |
最高研发阶段临床2期 |
首次获批国家/地区- |
首次获批日期- |
靶点- |
作用机制- |
|
|
|
非在研适应症- |
最高研发阶段早期临床1期 |
首次获批国家/地区- |
首次获批日期- |
/ Not yet recruiting早期临床1期IIT An Exploratory Clinical Study to Evaluate LX107 Gene Therapy in Patients With AIPL1 Biallelic Mutation-related Inherited Retinal Dystrophy (AIPL1-IRD)
Administering subretinal injection of LX107 injection (a gene therapy drug) to patients with retinal dystrophy caused by AIPL1 gene mutation to evaluate its efficacy and safety.
An Exploratory Clinical Study Evaluating LX111 Gene Therapy in Patients With Neovascular Age-related Macular Degeneration (nAMD)
The goal of this study is to evaluate the safety and efficacy of LX111 treatment of nAMD. This study will enroll subjects aged ≥ 50 vears old to receive a single unilateral intravitreal (lVT) injection of LX111 to evaluate its safety and efficacy.
/ Active, not recruiting临床2期 A Phase 2, Randomized Controlled, Open-Label Study to Establish the Safety and Efficacy of LX102 in Patients With Neovascular Age-related Macular Degeneration (nAMD) (VENUS)
The goal of this study is to evaluate the overall safety and efficacy of LX102 gene therapy for nAMD.
100 项与 上海朗昇生物科技有限公司 相关的临床结果
0 项与 上海朗昇生物科技有限公司 相关的专利(医药)
RPE65 基因突变引起的遗传性视网膜变性 (IRD) ,是一种致盲性眼科遗传疾病,通常在婴幼儿期就开始发病,视网膜和视觉功能逐步退化。几乎所有患者最终都会发展为完全失明,传统治疗方案对此束手无策。
昨晚(7月22日),中国国家药监局药品审评中心(CDE)官网最新公示,上海朗昇生物科技有限公司申报的LX101注射液拟纳入突破性治疗品种。用于治疗RPE65双等位基因突变相关的遗传性视网膜变性(IRD)患者。
LX101 注射液是一款以重组 AAV 基因载体 (rAAV) 为载体的基因疗法物。在特定遗传疾病的治疗策略中,可以减缓甚至停止光感受器退化的过程,保留与中央视觉功能有关的视锥细胞,甚至替换丢失的细胞从而提言患者的视觉功能,是眼科基因治疗的关键策略。
LX101注射液是朗信生物自主研发的一款以rAAV为载体的基因疗法。产品通过对RPE65基因编码序列优化设计,高效表达人源RPE65蛋白,补偿因该基因突变导致的蛋白功能缺失,眼内给药后可有效治疗RPE65双等位基因突变相关的遗传性视网膜变性(IRD)患者。在此前进行的研究者发起的临床研究中,LX101表现出良好的安全性,且已在多位患者中见到视力提高的疗效。
LX101眼用注射液是目前国内递交的首个先天性黑蒙基因治疗新药临床试验申请。先天性黑蒙LCA是一类发生最早、最严重的遗传性视网膜病变, 大多数LCA患者在婴儿期或儿童期开始出现严重视力障碍,并且由于进行性视网膜变性,在30-40岁时彻底失明。LCA目前尚无有效方法治疗,直到近些年来基因治疗领域的快速发展,这种严重视网膜遗传病才有了可用的治疗方法。
伴随全球基因治疗的快速发展,视网膜遗传病治疗领域近年来发生了革命性的变化。2017年12月10日,美国FDA批准了首个眼科基因治疗产品,通过腺相关病毒(AAV)载体,将正常的RPE65基因递送到视网膜细胞,用于治疗先天性黑矇症2型疗效甚佳,但其治疗费用高达85万美元,对于普通家庭来说无疑是“天价”,该药至今也未进入中国市场。
LX101注射液是一款以rAAV为载体的基因疗法,通过对RPE65基因编码序列优化设计,高效表达人源RPE65蛋白,补偿因该基因突变导致的蛋白功能缺失,眼内给药后可有效治疗RPE65双等位基因突变相关的遗传性视网膜变性(IRD)患者。
2021年6月,朗信生物启动了研究者发起的中国首项针对LCA的基因治疗临床研究,评估单次视网膜下腔注射LX101眼用注射液治疗特定类型先天性黑矇的总体安全性和疗效,于6月24日在上海市第一人民医院眼科中心成功完成首例患者治疗。
2021年9月16日,LX101完成了首位LCA患者的视网膜下注射,经过生物工程改造后,LX101带着目的基因片段进入由于基因突变而影响功能的细胞,在其中进行功能修复。
RPE65基因突变引起的遗传性视网膜变性(IRD),是一种典型的致盲性罕见病,常见的IRD包括视网膜色素变性、脉络膜症、Leber遗传性视神经病变(LHON)、Leber先天性黑矇(LCA)、Stargardt病、色盲症(ACHM)和X连锁视网膜劈裂症(XLRS)等,它们的发生是由于一种或多种基因的突变导致视网膜感光细胞死亡而引起。患者婴幼儿期发病,通常经历严重、进行性的视网膜退化和视觉功能恶化,几乎所有患者最终都会进展为完全失明。由于缺乏有效治疗方法,遗传性视网膜病变的致盲率居高不下,直到近些年来基因治疗领域的快速发展,这种严重视网膜遗传病才有了可用的治疗方法。
来源 | 北京药研汇
7月22日,中国国家药监局药品审评中心(CDE)官网最新公示,由朗信生物旗下上海朗昇生物申报的LX101注射液拟纳入突破性治疗品种,适用于治疗RPE65双等位基因突变相关的遗传性视网膜营养不良(IRD)患者。公开资料显示,LX101注射液是一款以rAAV为载体的基因治疗药物,其治疗RPE65突变相关IRD已进入3期临床试验阶段。
公开资料显示,RPE65基因突变引起的IRD是一种典型的致盲性罕见病,患者在婴幼儿期发病,几乎所有患者最终进展为完全失明。这类疾病临床尚无有效治疗方法。
LX101是上海朗昇生物研发的针对遗传性视网膜变性的基因治疗药物。该药物通过腺相关病毒2型(AAV2)携带正常RPE65基因,能特异识别并转染病变的视网膜色素上皮细胞,并在细胞内持续高效表达患者缺少的RPE65蛋白,恢复正常视循环,达到提升视功能的治疗目的。在此前进行的研究者发起的临床研究中,LX101表现出良好的安全性,且已在多位患者中见到视力提高的疗效。
公开资料显示,朗信生物成立于2020年,致力于基因治疗创新药物研发制造,专注于眼部遗传性和慢性疾病的基因治疗领域。今年6月,朗信生物宣布完成B+轮融资。该公司共推进5个候选分子进入临床,在3项致盲性眼病适应症中率先开展基因治疗临床研究,获得3项IND。除了LX101,治疗湿性年龄性黄斑变性(nAMD)的LX102、治疗X连锁视网膜劈裂症(XLRS)LX103均在推进研究。
参考资料:
[1]中国国家药监局药品审评中心(CDE)官网.Retrieved July 22,2024, From https://www.cde.org.cn/main/xxgk/listpage/da6efd086c099b7fc949121166f0130c
[2]【要闻】朗信生物LX101注射液I期临床试验完成首例患者给药. Retrieved July 28,2022, From https://mp.weixin.qq.com/s/iHWqYslzY-N9SsPdEIv8hA
内容来源于网络,如有侵权,请联系删除。
人类幻想世界中,战胜疾病、科幻进化的最终手段往往都来自“基因”,自1996年人类历史上第一只克隆生物克隆羊多利诞生之后,包括《千钧一发》《第六日》等电影作品的推出 ,让基因技术类科幻片迎来 了属于自己的小高潮,也让大众对基因技术这个全新技术有了意想层面的认识。
但谁曾想,短短十数年间的科学技术发展,基因疗法早已不再是想象中的技术,而是逐渐走入了我们的现实生活,开始服务于各种“罕见病新药”研发。
有望依靠基因疗法解决
据参考资料显示,约80%罕见病是源于基因缺陷而导致的遗传病,其中单基因病比例最高。而所谓基因疗法,是指利用载体将遗传物质引入靶细胞,通过纠正或补充缺陷基因来治疗或预防疾病,而该项技术的最佳应用场景又正是单基因疾病。
两者匹配之下,基因疗法与罕见病恰好相得益彰,基因疗法成为了罕见病治疗的全新力量。另外,由于在目前已知的7000多种罕见病中,超过95%的疾病并无FDA批准的针对性药物,常规治疗方法多是对症治疗与替代治疗,但这些方法只能缓解症状,并不能根治病因,且可能还伴随一定程度的副作用和风险。如血友病的凝血因子治疗、DMD的糖皮质激素治疗等等。所以,最终导致罕见病临床上对病因调控类型药物的需求长期存在,相关的支付意愿也相对较高。
基因疗法的出现,不同于传统小分子药物和抗体药物在蛋白质水平进行调控,其可以在DNA或mRNA水平上对致病基因进行修正从而达到治疗效果,既克服传统小分子和抗体的不足,对于致病基因清晰的罕见病几乎可以做到,将症状治疗完全转变成为病因治疗,一劳永逸,应用空间巨大,对于罕见病治疗更是具有“划时代”意义。
治疗领域为罕见病
虽不能说基因疗法只能应用于罕见病领域,毕竟肿瘤、代谢、慢病领域已经开始出现了基因疗法的具体应用,未来的基因疗法甚至大概率将与生活中的常规疾病结合,比如前段时间,Fractyl Health展示的其“Rejuva基因治疗平台”的临床前研究成果,利用腺相关病毒(AAV)载体实现“一次给药持久减重”,单剂输注后28天,将小鼠体重降低27%,高于了对照组持续司美格鲁肽治疗的21%数据。
但是,以目前基因疗法的治疗领域分布来看,非罕见病领域所占的比例有限,目前仍主要以罕见病与罕见肿瘤亚型为主,在所有全球985项基因疗法研究中,罕见病治疗领域有485项,占比49.23%,这还是不包含罕见病肿瘤目录中罕见肿瘤亚型的基础上,如果将罕见肿瘤归入其中,占比还会进一步提升。
同时,由于罕见病遗传特性,导致绝大多数罕见病会在儿童阶段发病,儿童病症表现明显,而无论FDA还是CDE,对于儿童罕见病都拥有各自的突破性疗法机制、有限审评机制。对于药企,尤其是部分biotech企业而言,选择儿童罕见病适应症一定程度上可以享受管线加速的优势,积极性大幅上涨。
适应症为“罕见病”
2023年,美国FDA批准新药中,包含了5款全新的基因疗法,且其中4款亦选择了罕见病领域,分别是针对镰状细胞病(SCD)的Casgevy与Lyfgenia、针对A型血友病的Roctavian以及针对杜氏肌营养不良症(DMD)的Elevidys;
2017至今,历史获批的10款基因疗法中,更是有9款是针对的罕见病领域;
并且,随着全球医学界对基因治疗的重视程度加深,以及科学水平的持续上涨,越来越多值得庆贺的成果面世,极大地鼓励了医药研发的热情。
2012年,欧洲药品管理局EMA批准了全球首款AAV疗法Glybera上市;
2016年,FDA发布6大新指南,全力推动基因治疗;
2017年,FDA才首次向AAV疗法敞开上市之门,诺华Luxturna被批准用于双等位基因 RPE65 突变相关性视网膜营养不良;
2022年,FDA首次批准了3款基因疗法上市,验证了全力推动基因治疗的最初设想;
2023年,FDA全年共批准了5款基因疗法,创历史新高;
当罕见病新药临床需求越来越迫切,当基因治疗药物获批数量一年比一年多,当罕见病与基因治疗的匹配度越来越高,罕见病治疗或许也将借此开启一个全新的基因治疗时代。
基因回补与基因编辑同步进行
而一般情况下,罕见病的基因治疗大致策略可分为两者,一者是针对由特定基因缺陷引起的疾病,可通过递送缺失功能基因的正常版本来纠正,简称“基因回补”;二者是针对由错误折叠的毒性蛋白引起的疾病,可以通过递送基因编辑工具对突变基因进行敲除或纠正,简称“基因编辑”。
在体基因治疗的两种策略
目前,基因回补最常见的即“基因递送技术”,常用的病毒载体包括腺相关病毒载体、慢病毒载体和腺病毒载体三种,各自均有相对应的优缺点,腺病毒载体(AVV)应用较广泛,也是最有望为罕见病患者带来根治性新药的一种。至于基因编辑则目前主要作为部分致病基因过大,超出病毒载体载限的进阶技术,近年来发展迅速。
罕见病基因疗法应用最广泛
凭借基因疗法在部分领域高转导效率等因素,据药智数据显示,79项目活跃管线(近一年有临床进展)中,血液系统、神经系统、肌肉骨骼与眼科罕见病领域是目前基因治疗应用最多的领域。
在FDA已获批的10种基因疗法中,有一半是针对的血液系统疾病,包括Roctavian(血友病A)、Hemgenix(血友病B)、Zynteglo(β-地中海贫血)、Lyfgenia(SCD)与Casgevy(TDT/SCD),基因递送与基因编辑两类均有涉及。
其中,最值得注意的是,FDA同一天获批,针对同一适应症的两款不同的新药Lyfgenia与Casgevy,一款采用的是传统基因递送技术,另一个却是全球首个CRISPR基因编辑疗法(曾经的诺贝奖得奖技术),完全不同的两种策略方式被FDA放在一起对比,既是对两种技术的认可,也展现了其多基因疗法的开明态度。
Lyfgenia:使用慢病毒将增强型β珠蛋白嵌入患者自身造血干细胞基因组中,持久产生具有抗镰状细胞特性的血红蛋白(HbAT87Q),从而减少VOE的发生。
Casgevy:是利用CRISPR技术对BCL11A基因敲除,使得造血干细胞中γ珠蛋白的表达上调,β珠蛋白表达下降,从而产生胎儿血红蛋白HbF。
据参考资料内容显示,大约60%的单基因病最终都会导致神经系统症状,截止目前很多引发神经系统疾病的基因突变类型基本得以确认。
而基因载体递送技术,可通过一次给药将治疗基因转入神经元或其他神经细胞中,且具有长期疗效,在中枢神经系统疾病的治疗中有很大的优势,如利用AAV递送到帕金森病患者大脑中的芳香氨基酸脱羧酶就可持续表达4年,而在非人灵长类动物中则可持续表达15年。
在FDA批准的10款基因疗法中,2022年获批用于活动性脑肾上腺脑白质营养不良的SKYSONA就是唯一一款用于神经系统疾病的基因疗法,其利用Lenti-D慢病毒载体(LVV)进行体外转导,将ABCD1基因的功能拷贝添加到患者自身的造血干细胞(HSC)中,从而尽可能地保留神经功能,包括保留运动功能和沟通能力。
在罕见病领域中有很大一部分的疾病属于神经肌肉病,其主要影响神经肌肉单元的一个或多个组成部分,包括运动神经元和骨骼肌两个部分。最显著的例子就是杜氏肌营养不良症和脊髓性肌肉萎缩症。
同时,由于这类神经肌肉疾病大多具有明确的遗传缺陷和单基因病性质,使得基因治疗成为该领域被寄予厚望的治疗方法。
以I型脊髓性肌萎缩(SMA)为例,其是SMN 1基因异常所致的新生儿急性致命性神经肌肉疾病中,利用可穿越血脑屏障的AAV 9载体,以静脉注射的方式将携带的SMN 1基因递送给SMA婴幼儿,使受试患儿的生存时间大大延长,运动功能也得到改善。
2019年批准的Zolgensma与2023年批准的Elevidys,就是FDA唯二批准用于神经肌肉罕见病治疗的基因疗法。
据参考资料显示,生理状态下,血视网膜屏障和血房水屏障2套血眼屏障的存在使得眼睛的免疫系统相对独立,因此采用视网膜下注射AAV的方式局部递送目的基因入细胞,可有效避免抗原诱发的炎症免疫反应,是基因治疗视网膜及视神经疾病的一大优势。
2017年,FDA历史第一款获批上市的基因治疗药物Luxturna,就是针对的双等位基因 RPE65 突变相关性视网膜营养不良,其通过视网膜下注射的方式递送AAV载体中的人类RPE65基因的正确编码序列到视网膜色素上皮,实现真正意义上的“重见光明”。
国内罕见病基因疗法研发火热
与海外相比,我国罕见病基因治疗起步较晚,现阶段罕见病基因疗法管线仅有美国的1/3左右,且多数基因疗法尚处于临床前期,但由于我国较大的患者基数,大量基因疗法基础研究的进步与相关人才爆发,我国罕见病基因治疗的发展速度远高于全球平均水平,截至目前,基因疗法的临床试验数量仅次于美国,增速全球第一。
政策上,国家药品监督管理局已经针对基因疗法形成了由法律法规、管理制度与指南文件组成的初步完善的审批流程和监管体系,且药品审评中心还多次发布了基因治疗的相关指导文件,例如2021年12月的《罕见疾病药物临床研发技术指导原则》等,对国内基因治疗的支持态度明显,推动作用巨大。
药品研发方面,据药智数据显示,截至目前国内原研的基因疗法共有80余款管线进入了临床阶段,其中近一年活跃的临床III期管线有3款,分别是朗昇生物的LX-101、诺思兰德生物的VM-202、信致医药的BBM-H901,活跃II期管线则相对较多,共计14款,其中绝大多数适应症均围绕罕见病与眼科疾病布局,基本符合基因治疗的根本特性,如包括血友病、β地中海贫血以及老年性黄斑变性。
近一年国内原研活跃基因疗法(部分)
按照目前国内基因治疗的发展速度,很难想象未来5-10年,会有多少相关研究得以落地,相关药品得以上市,赶超海外市场或许并非遥不可及。
基因治疗作为一种新兴的治疗手段,原则上有望解决所有单基因病的治疗困境,而作为单基因病集中地的罕见病领域则也最有望取得率先突破。
目前来讲,基因递送技术已经被应用于多项罕见遗传病的临床试验,涵盖眼病、血液病、神经系统疾病等多个领域,AAV作为当下最安全、长效的病毒载体,是治疗罕见单基因病最被寄予厚望的平台,有望在未来5—10年里全面爆发。另外,基因编辑技术的应用愈发熟练,技术走向了成熟,也使原有因致病基因过大,超出病毒载体的包装限制而不能运用基因递送方法治疗的疾病得到了突破性进展,首款基因编辑疗法Casgevy获批上市不仅拉开了基因治疗的新序幕,后续TALEN/ZFN/碱基编辑等基因编辑技术也在迅速发展中,前途不可限量。
但是,基因治疗作为一种复杂疗法,要想应用至临床来治疗更多更广泛的遗传性罕见病,尚存在诸多问题亟待解决,如改善整合基因载体和脱靶效应带来的遗传毒性、控制靶基因体内的表达量维持在有效治疗疾病水平、在不降低其活性的基础上延长半衰期、解决伦理问题和高昂的治疗费用等。
在这些方面,全球基因疗法的研发企业和CDMO企业也在积极布局,从研发与技术上、从工艺端,应对工艺难点,降低工艺成本和偏差,如博腾股份旗下的博腾生物,作为细胞与基因治疗CRO+CDMO公司,可以提供基因编辑CRO服务与质粒和AAV相关CDMO服务,已投入运行的10条病毒载体线可完全满足基因治疗产品的临床期与商业生产需要。
前路漫漫亦灿灿,唯愿更多有识之士能共同努力,为更多罕见病患者的基因治疗需求奋斗终生,让更多幻想能成为现实。
参考资料:
宣建伟, 孙巧. 中国罕见病药物经济学评估适用模型与支付阈值参考标准探讨[J]. 国际药学研究杂志, 2019(9).
李玉欢,李伟.罕见病的基因治疗研究进展.《罕少疾病杂志》,2023(3)
桂怡婷,李强 ,桂永浩.罕见病的基因治疗应用与展望.《临床儿科杂志》. 2020(10)
基因疗法在罕见病上的应用, 劲帆生物,2023-03-31
百亿罕见病市场,基因疗法或为破局之道, 同写意, 2023-04-12
内容来源于网络,如有侵权,请联系删除。

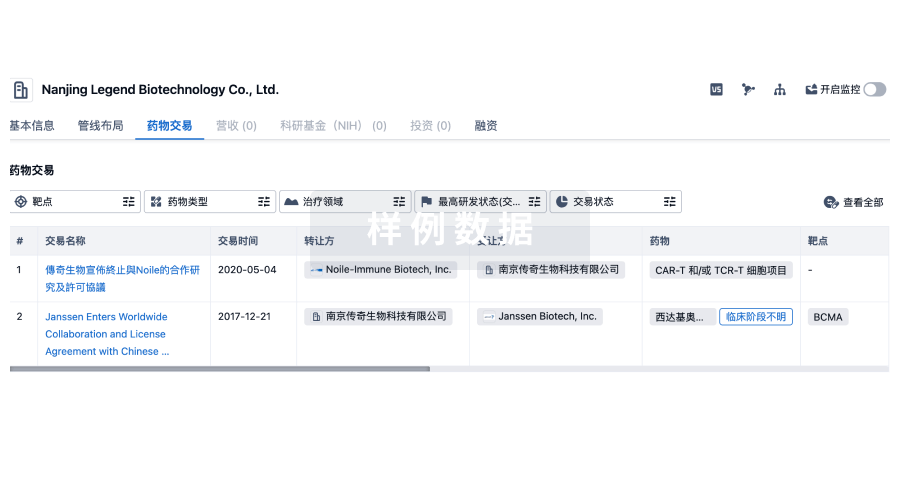
100 项与 上海朗昇生物科技有限公司 相关的药物交易
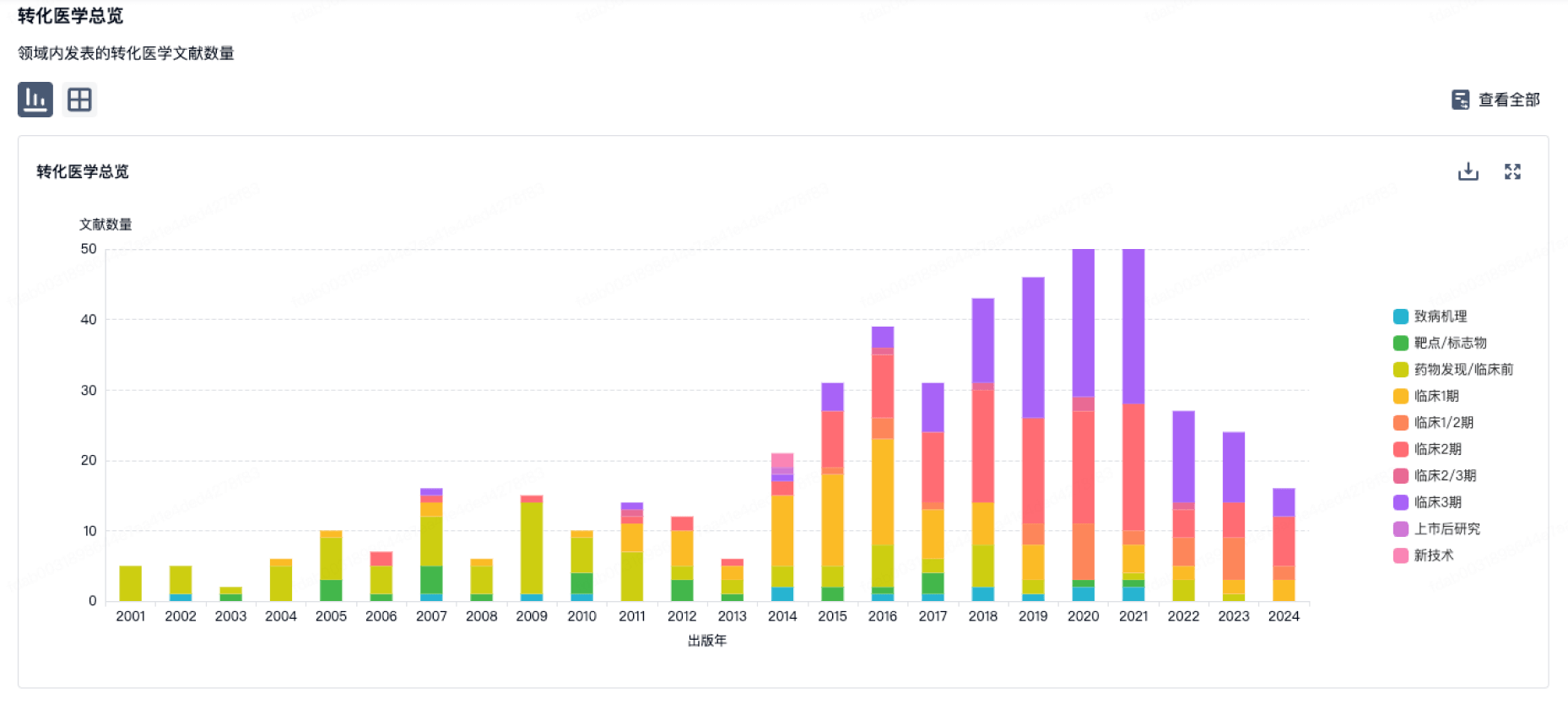
100 项与 上海朗昇生物科技有限公司 相关的转化医学