|
|
|
|
|
非在研适应症- |
最高研发阶段批准上市 |
|
首次获批日期2023-01-06 |
|
|
|
|
|
|
最高研发阶段临床2期 |
首次获批国家/地区- |
首次获批日期- |
|
|
|
|
|
非在研适应症- |
最高研发阶段临床前 |
首次获批国家/地区- |
首次获批日期- |
A Phase 2a, Randomized, Double-blind, Placebo-controlled Trial to Evaluate the Safety, Tolerability, and Pharmacokinetics of Multiple Ascending Dosing of Exidavnemab in Patients With Mild to Moderate Parkinson's Disease on Stable Symptomatic Parkinson's Disease Medication and in Patients With Multiple System Atrophy
The primary objective of this study is to assess the safety and tolerability of exidavnemab after multiple dosing versus placebo.
/ Not yet recruiting临床1/2期 An Open, Randomized, Rehabilitation-controlled Study to Assess Safety, Tolerability, and Efficacy of Heparin Activated Recombinant Human Fibroblast Growth Factor 1 on a Biodegradable Device in Subjects with Traumatic Spinal Cord Injury
An Open, Randomized, Rehabilitation-controlled Study to Assess Safety, Tolerability, and Efficacy of Heparin Activated Recombinant Fibroblast Growth Factor 1 on a Biodegradable Device in Subjects With Traumatic Spinal Cord Injury
This is an open, randomized, rehabilitation-controlled study in subjects with complete Traumatic Spinal Cord Injury, where the active treatment consists of a surgical implantation of SC0806 (a biodegradable device with heparin-activated FGF1 and nerve implants).
100 项与 BioArctic AB 相关的临床结果
0 项与 BioArctic AB 相关的专利(医药)
氨基观察-创新药组原创出品
作者 | 黄恺
多家明星药企,带来亮眼成绩单。
8月26日,映恩生物公告,上半年总营收12.29亿元,调整后利润1.46亿元,现金总额达到37.47亿元。
康诺亚也是在同一天宣布,上半年营收4.99亿元,同比暴增812%,其中包括合作收入3.29亿元。研发支出3.60亿元,同比增长9%。
礼来口服GLP-1一项3期临床成功。
8月26日,礼来公布了其在研GLP-1受体激动剂orforglipron的3期临床试验ATTAIN-2的积极顶线结果。该研究针对合并2型糖尿病的肥胖或超重成人患者。在ATTAIN-2研究中,orforglipron的3个剂量组均达到了主要终点和所有关键次要终点,在72周时实现了显著的体重下降、具有临床意义的糖化血红蛋白(A1C)降低,及心血管风险因素的改善。在主要终点方面,每日一次且在不受食物或饮水限制的前提下,orforglipron 36mg组平均体重下降达10.5%(10.4kg),而安慰剂组为2.2%(2.3kg)。
在过去的一天里,国内外医药市场还有哪些热点值得关注?让氨基君带你一探究竟。
/ 01 /
市场速递
1)9.5亿美元出海,百济改写BD交易逻辑
8月25日,百济神州宣布与Royalty Pharma签订特许权使用费购买协议。后者支付8.85亿美元首付款,获得百济神州旗下塔拉妥单抗(Imdelltra,tarlatamab)在中国以外地区销售的特许权使用费权益。根据协议条款,百济神州有权在12个月内出售剩余的收取特许权使用费的权利,由此最高可获得6500万美元的额外付款。百济神州还将根据特许权使用费所占比例,享有该产品年销售额超15亿美元的部分收入。
2)汇宇制药拟与同源康签署全国总代理协议
8月26日,汇宇制药公告,全资子公司汇辰昕拟与同源康签署《关于同源康“TY-9591”产品的全国总代理协议》,同源康同意将其“TY-9591”产品的全国总代理权授权给汇辰昕,里程碑首付款为15000万元人民币。“TY-9591”产品是由同源康自主研发的可口服、不可逆的第三代EGFR(表皮生长因子受体)抑制剂。
/ 02 /
资本信息
1)君实生物上半年营业收入同比增长48.64%
8月26日,君实生物公告,2025年上半年公司营业收入为11.68亿元,同比增长48.64%;归属于上市公司股东的净亏损4.13亿元,上年同期净亏损6.45亿元。
2)复星医药上半年净利润同比增长38.96%
8月26日,复星医药公告,2025年上半年营业收入195.14亿元,净利润17.02亿元,同比增长38.96%。
3)映恩生物上半年调整后利润1.46亿元,现金37.47亿元
8月26日,映恩生物公告,上半年总营收12.29亿元,调整后利润1.46亿元,现金总额达到37.47亿元。
4)康诺亚上半年营收暴增812%
8 月 26 日,康诺亚发布2025上半年业绩公告,营收4.99亿元,同比暴增812%,其中包括合作收入3.29亿元。研发支出3.60亿元,同比增长9%。
/ 03 /
医药动态
1)康诺亚CM336注射液获临床许可
8月26日,据CDE官网,康诺亚CM336注射液获临床许可,拟用于自身免疫性血细胞减少症。
2)百济神州Sonrotoclax薄膜包衣片获临床许可
8月26日,据CDE官网,百济神州Sonrotoclax薄膜包衣片获临床许可,拟联合抗CD20抗体治疗复发和/或难治性慢性淋巴细胞白血病/小淋巴细胞淋巴瘤。
/ 04 /
海外药闻
1)礼来口服GLP-1RA orforglipron肥胖合并2型糖尿病的3期临床研究成功
8月26日,礼来公布了其在研GLP-1受体激动剂orforglipron的3期临床试验ATTAIN-2的积极顶线结果。该研究针对合并2型糖尿病的肥胖或超重成人患者。在ATTAIN-2研究中,orforglipron的3个剂量组均达到了主要终点和所有关键次要终点,在72周时实现了显著的体重下降、具有临床意义的糖化血红蛋白(A1C)降低,及心血管风险因素的改善。在主要终点方面,每日一次且在不受食物或饮水限制的前提下,orforglipron 36mg组平均体重下降达10.5%(10.4kg),而安慰剂组为2.2%(2.3kg)。
2)诺华押注神经退行性疾病新疗法
8 月 26 日,BioArctic AB (publ) 宣布,已与诺华达成一项期权、合作及许可协议。该协议将 BioArctic 专有的 BrainTransporter 技术与神经退行性疾病领域一个未公开的靶点相结合,开发一种潜在的新疗法。作为初步研究合作的一部分,BioArctic 将获得 3000 万美元的预付款。
PS:欢迎扫描下方二维码,添加氨基君微信号交流。
2025年8月26日,瑞典生物技术公司BioArctic AB宣布与诺华达成一项期权、合作及许可协议。
该协议将 BioArctic 专有的 BrainTransporter 技术与神经退行性疾病领域一个未公开的靶点相结合,开发一种潜在的新疗法。诺华将评估初始合作期间产生的数据,并决定是否行使选择权来授权任何由此产生的候选药物。如果诺华行使选择权,它将承担全球开发和商业化的全部责任。
根据协议条款,BioArctic有资格获得3000万美元的预付款。如果诺华行使选择权,BioArctic还有资格获得高达7.72亿美元的额外里程碑付款,以及未来全球产品销售额的分级特许权使用费。
BioArctic的BrainTransporter技术是一种利用转铁蛋白受体(TfR)促进生物药物(例如抗体)进入脑部的技术平台。该技术通过主动运输生物治疗药物穿过血脑屏障,可以使药物在脑内分布更广,从而提高疗效、改善安全性并增强给药便利性。这项技术正在应用于多个内部药物项目,并可能成为未来与其他制药公司合作的一部分。BrainTransporter技术可以用于多种治疗领域的生物制剂和其他模式的递送。
BBB:血脑屏障
RMT:受体介导的运输
关于 BioArctic BioArctic AB (publ) 是一家瑞典研究型生物制药公司,专注于可以延缓或阻止神经退行性疾病进展的创新疗法。该公司发明了 Leqembi®(lecanemab)——世界上第一种被证明可以减缓疾病进展并减少早期阿尔茨海默病认知障碍的药物。Leqembi 是与 BioArctic 的合作伙伴卫材共同开发的,卫材负责全球监管互动和商业化。除了 Leqembi 之外,BioArctic 还拥有广泛的研究组合,包括针对帕金森病和 ALS 的抗体以及针对阿尔茨海默病的其他项目。其中一些项目利用了该公司专有的 BrainTransporter™ 技术,该技术有可能主动转运抗体穿过血脑屏障,以提高治疗效果。
End
声明:本公众号所有发文章(包括原创及转载文章)系出于传递更多信息之目的,且注明来源和作者。本公众号欢迎分享朋友圈或大群,谢绝媒体或机构未经授权以任何形式转载至其他平台。
转载/商务/投稿 | 联系微信15618157102(sum_Gmi)
商务合作
稿件征集
点击了解详情
往期回顾
1
港股创新药走出至暗时刻
2
License-out潮来袭:谁是下一个出海爆款?
3
高盛发声:中国创新药价值重估时代来临
当地时间 8 月 26 日,BioArctic AB (publ) 宣布,已与诺华达成一项期权、合作及许可协议。
截图来源:企业官网
该协议将 BioArctic 专有的 BrainTransporter 技术与神经退行性疾病领域一个未公开的靶点相结合,开发一种潜在的新疗法。
BrainTransporter 是一种利用转铁蛋白受体 (TfR) 促进生物药物(例如抗体)进入脑部的技术。生物治疗药物主动转运穿过血脑屏障,可以使其在脑内分布更广,从而提高疗效、安全性和给药便利性。
作为初步研究合作的一部分,BioArctic 将获得 3000 万美元的预付款。
此外,诺华将评估初始合作期间产生的数据,并决定是否行使选择权,授权任何候选药物。如果诺华行使选择权,BioArctic 将有资格获得高达 7.72 亿美元的额外付款。如果产品上市,BioArctic 还将有权获得未来全球产品销售额的分级个位数特许权使用费。
根据初步研究合作,BioArctic 将结合 BrainTransporter 技术与诺华专有抗体,开发一种新的候选药物。如果诺华在评估该候选药物后决定行使选择权,诺华将全权负责该候选药物及相关产品的全球开发和商业化。
识别微信二维码,可添加药时空小编
请注明:姓名+研究方向!
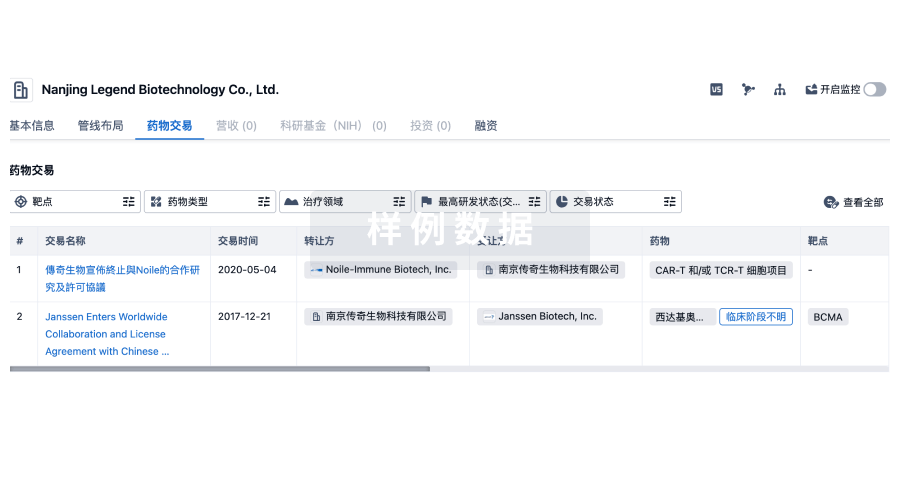
100 项与 BioArctic AB 相关的药物交易
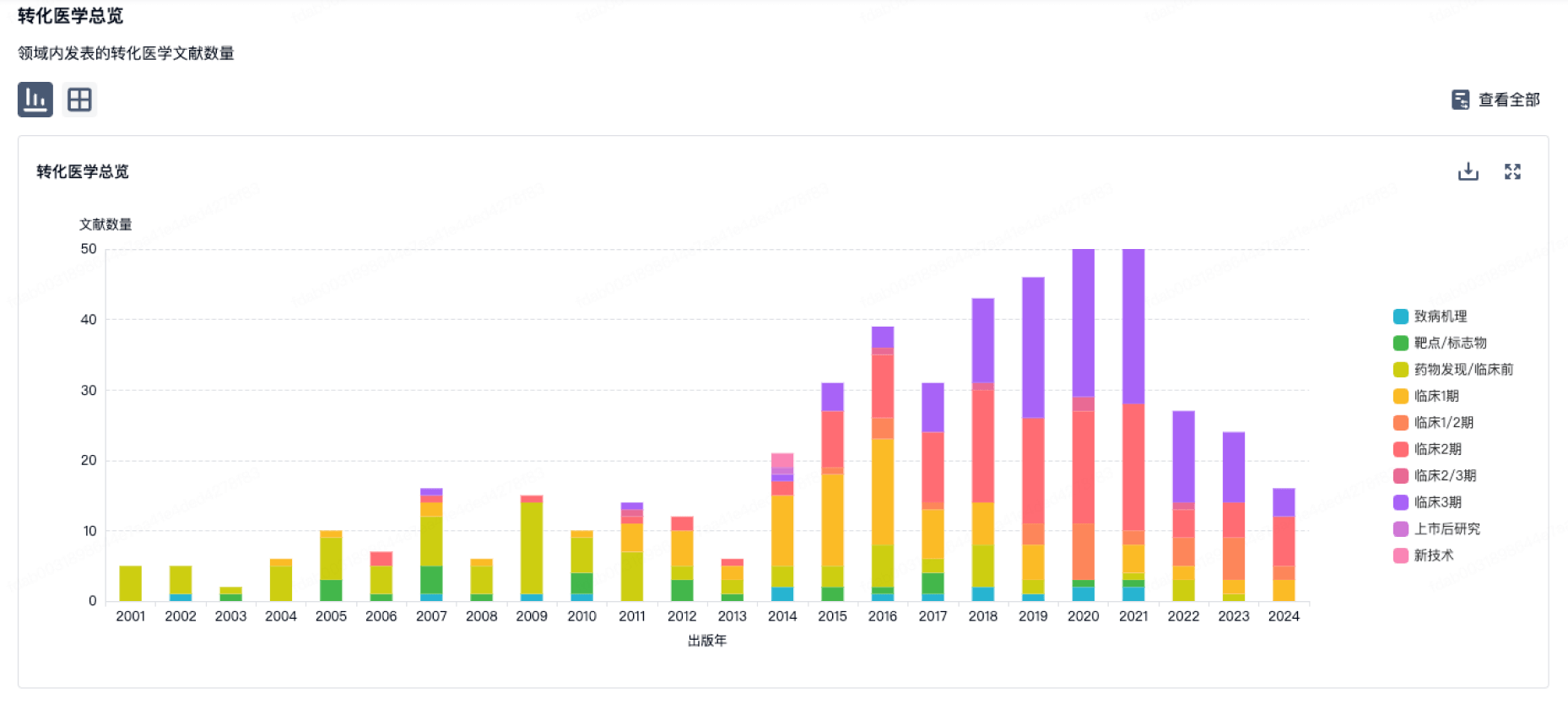
100 项与 BioArctic AB 相关的转化医学