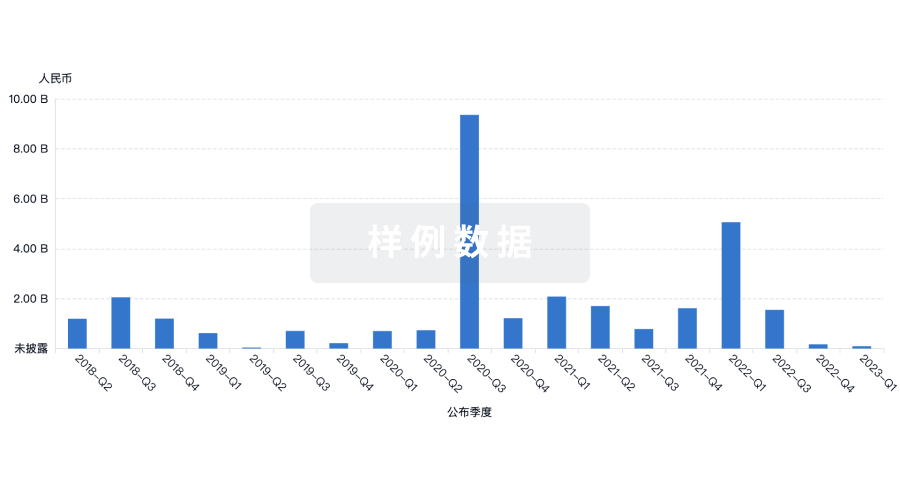
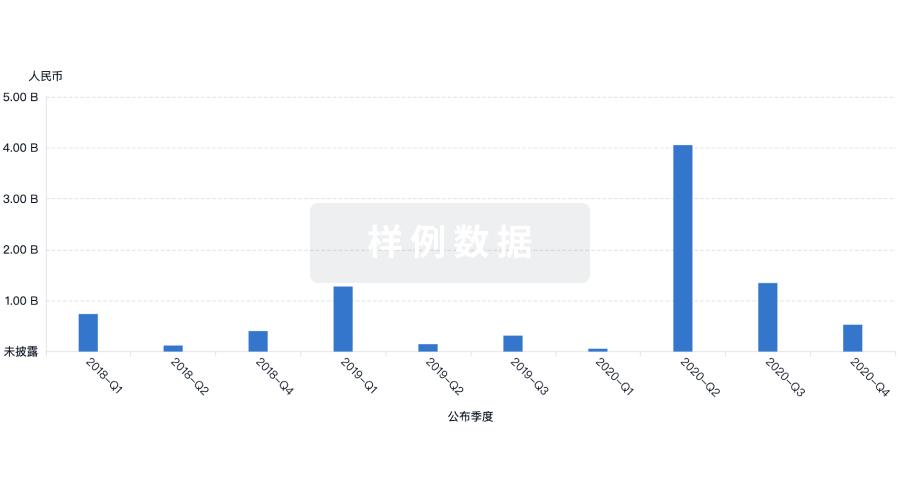
靶点- |
作用机制- |
|
|
|
非在研适应症- |
最高研发阶段批准上市 |
|
首次获批日期1987-05-05 |
|
|
|
|
|
非在研适应症- |
最高研发阶段早期临床1期 |
首次获批国家/地区- |
首次获批日期- |
靶点- |
|
|
|
|
非在研适应症- |
最高研发阶段早期临床1期 |
首次获批国家/地区- |
首次获批日期- |
Research on Intraoperative Hypothermia Risk Prediction and Temperature Management Strategies in Elderly Patients: Construction of Intraoperative Hypothermia Prediction Model Based on Dynamic Incremental Training and Evaluation of Clinical Application Effectiveness
With the support of partial dual temperature monitoring (comparing the specific difference between standardized axillary temperature monitoring and esophageal temperature), this trial is divided into the following three parts:
1. Multi center observational study: Establish and validate a dynamic incremental training intraoperative hypothermia prediction model - Intelligent Care for the Elderly (ICE) - Intraoperative hypothermia warning system, and provide ICE Offline for use by healthcare professionals and ICE Online for further model updates when needed for clinical or research purposes.
2. Multi center non randomized controlled clinical trial: Conduct a multi center stratified temperature management clinical trial based on ICE Offline after dynamic incremental training to verify the clinical and economic benefits of the model and active warming.
3. Pre and post comparative study: Collect data before ICE application and compare it with data after ICE promotion.
A Single-blind Randomized Controlled Trial to Evaluate Efficacy and Safety of Immersive Virtual Reality for Post-stroke Cognitive Impairment
This is a single-blind randomized controlled trial to evaluate the efficacy and safety of immersive virtual reality for post-stroke cognitive impairment.
Effectiveness of Real Home Settings Via Virtual Reality Task Oriented Training on Upper Limb Function in Patients with Stroke
Stroke rank second among the top causes of death, affecting millions of people in the worldwide. It has been reported that hemiplegia is the most common sequelae after stroke, accounting for about 50%-70% of all sequelae of the disease. About 75% of stroke patients are accompanied by different degrees of upper limb dysfunction, which seriously affects the activities of daily life and cause serious physical and mental burden to patients and their families. Early recovery of upper limb motor function is a great significance for the overall recovery of stroke patients. Task-oriented training (TOT) is reported to improve the motor coordination and ADL. However, lack varies of tasks limited the treatment ability for patients with stroke hemiplegia during hospital admission. Virtual reality (VR) offers advantages of providing virtual scenes that is difficult in the real world, such as the scene of garden, camara, and plaza etc. And the familiar circumstances for patients may have the potential to increase the motivation of rehabilitation training, and improve the efficacy of occupational therapy (OT).
The goal of this study is to observe the effectiveness of real home settings via virtual reality assisted TOT on upper limb function in patients with stroke. Functional near-infrared spectroscopy (fNIRS) and electroencephalography (EEG) were used to observe the changes in brain function under VR-TOT training.
We intended to recruit 120 participants, and allocate to three groups: VR-TOT, TOT, and traditional OT. Each of them completed the Fugl-Meyer-UE, Wolf motor function test (WMFT), hand gripping power, modified Ashworth、Purdue Pegboard test (PPT)、modified Barthel index (MBI)、mini mental state examination (MMSE)、NIH stroke scale (NIHSS)、Virtual reality sickness questionnaire (VRSQ), Intrinsic Motivation Inventory Inventory (IMI), satisfaction VAS, body representation, sense of ownership, Proprioceptive Drift scale before and after the treatment. Additionally, we conducted fNIRS and EEG at baseline and during the follow up to understand the changes in brain function.
100 项与 Chongqing Medical University /The First Affiliated Hospital/ 相关的临床结果
0 项与 Chongqing Medical University /The First Affiliated Hospital/ 相关的专利(医药)
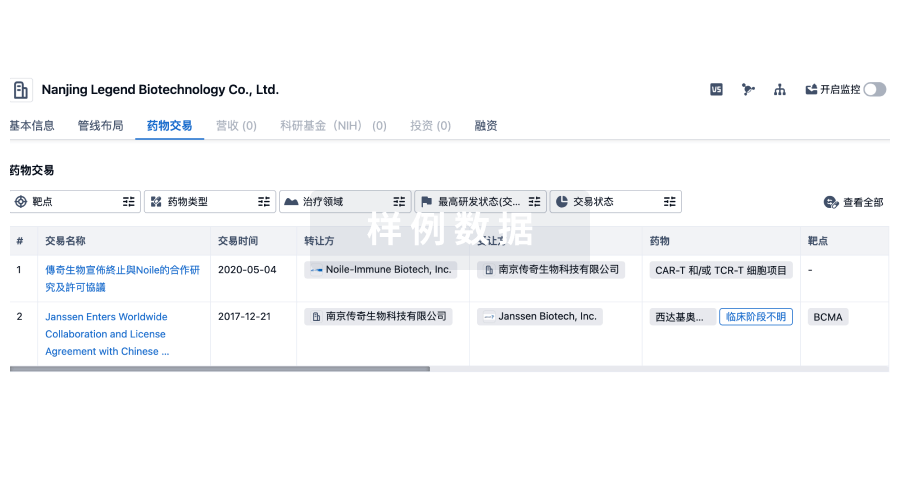
100 项与 Chongqing Medical University /The First Affiliated Hospital/ 相关的药物交易
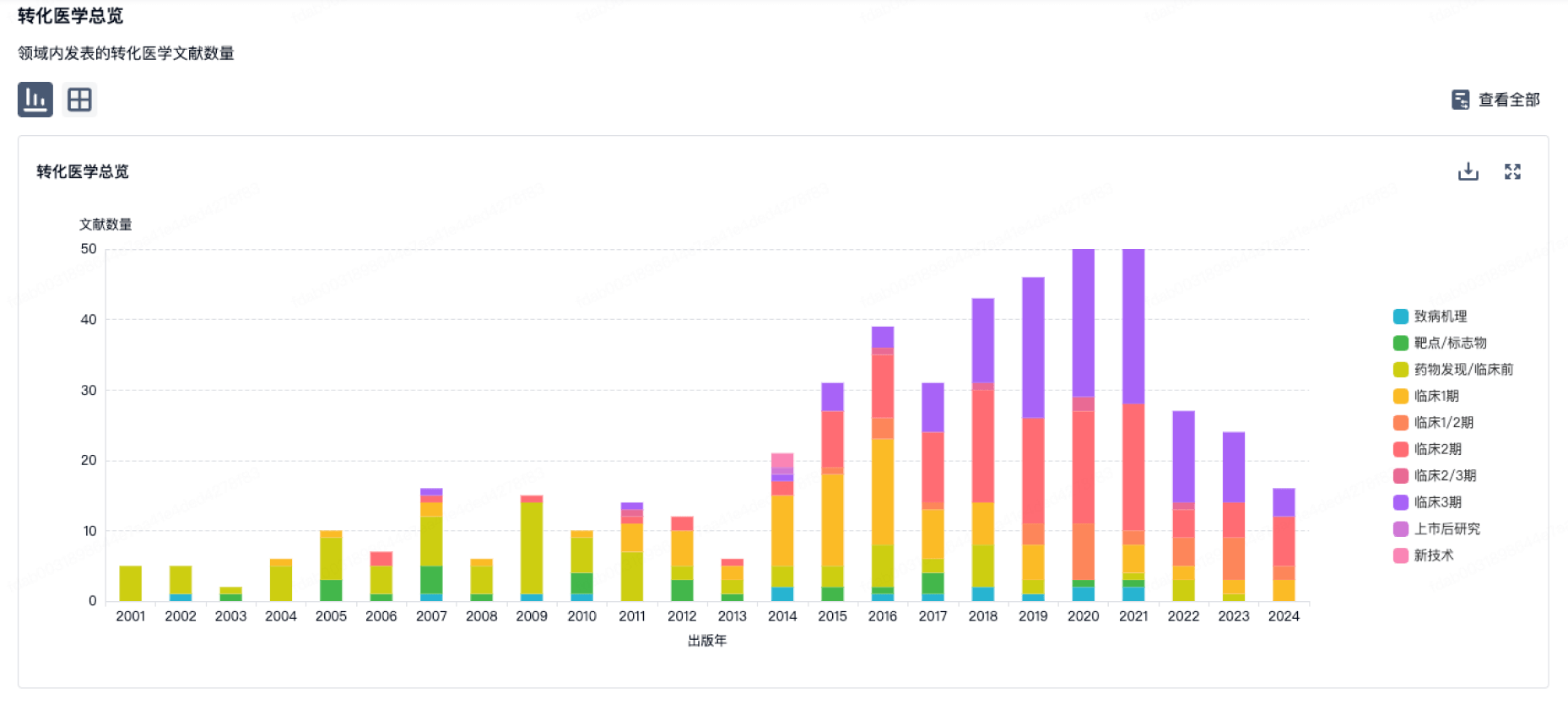
100 项与 Chongqing Medical University /The First Affiliated Hospital/ 相关的转化医学