预约演示
更新于:2025-05-07
Wuxi Zhikang Hongren New Drug Development Co., Ltd.
注销
| 无锡智康弘仁新药开发有限公司|中国注销
| 无锡智康弘仁新药开发有限公司|中国更新于:2025-05-07
概览
标签
呼吸系统疾病
其他疾病
小分子化药
疾病领域得分
一眼洞穿机构专注的疾病领域

暂无数据
技术平台
公司药物应用最多的技术
暂无数据
靶点
公司最常开发的靶点
暂无数据
| 排名前五的药物类型 | 数量 |
|---|---|
| 小分子化药 | 1 |
关联
1
项与 无锡智康弘仁新药开发有限公司 相关的药物靶点- |
作用机制- |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段临床2/3期 |
首次获批国家/地区- |
首次获批日期- |
2
项与 无锡智康弘仁新药开发有限公司 相关的临床试验CTR20230327
一项随机、双盲、安慰剂对照评价舒非尼酮(SC1011)片在特发性肺纤维化(IPF)患者中的有效性和安全性的II/III期临床研究
主要目的:评价舒非尼酮(SC1011)片治疗IPF的疗效
次要目的:评价舒非尼酮(SC1011)片治疗IPF患者的其他疗效
开始日期- |
申办/合作机构 |
CTR20211374
研究评估SC1011片对健康成人志愿者的耐受性、药代动力学特征
主要目的:评估健康成人单次剂量递增和多次剂量递增口服SC1011片的安全性和耐受性。
次要目的:评估进食对健康成人单次口服SC1011片药代动力学的影响。
开始日期2021-05-27 |
申办/合作机构 |
100 项与 无锡智康弘仁新药开发有限公司 相关的临床结果
登录后查看更多信息
0 项与 无锡智康弘仁新药开发有限公司 相关的专利(医药)
登录后查看更多信息
4
项与 无锡智康弘仁新药开发有限公司 相关的新闻(医药)2024-06-19
·药时代
近日,据报道,在两名受试者死亡后,FDA停止了Zentalis公司包括ZN-c3-001、Denali、TETON在内的三项研究。
受此消息影响,Zentalis股价在周二的盘前交易中暴跌约30%。同时,包括Wedbush的分析师Robert Driscoll、奥本海默(Oppenheimer)分析师Matthew Biegler、富国银行、Stifel、HC Wainwright、摩根士丹利、Jefferies均下调了对Zentalis评级。
01
事件全貌
被FDA叫停临床研究的药物是Zentalis的一款Wee1抑制剂——Azenosertib。
简单介绍一下Wee1。
Wee1是DNA损伤修复通路中重要的调控激酶,属于Wee蛋白激酶家族,在多种肿瘤细胞中高度表达。研究发现,当Wee1激酶被抑制时,肿瘤细胞细胞周期进程加速,DNA损伤持续累积,最终细胞分裂受阻,进一步导致肿瘤细胞凋亡。
图片来源:Zentalis官网
Wee1抑制剂是继PARP抑制剂在临床获得成功后,DNA损伤修复通路中的又一热门靶点,具有开发成为新一代靶向疗法的潜力。同时,除单药治疗外,Wee1抑制剂还在联合用药展现出了良好前景,比如与放化疗、ATR抑制剂、免疫治疗药物等联合用于多种肿瘤的治疗。
根据Zentalis官网信息,三项被FDA停止的研究分别为:
1)TETON:Azenosertib单药治疗子宫浆液性癌( Uterine Serous Carcinoma,USC)的研究;
2)Denali:Azenosertib单药治疗高级别浆液性卵巢癌(Cyclin E1 Driven High Grade Serous Ovarian Cancer)的研究;
3)ZN-c3-001:Azenosertib单药治疗实体瘤。
不难看出,此次被FDA叫停的三项研究均属于WEE1抑制剂单药治疗方案。在半个月前的ASCO年会上,Zentalis CEO Kimberly Blackwell博士曾表明,在近一两年将得出关于azenosertib单一疗法的活性、安全性及有效性相关数据。
而在试验被FDA叫停后,Zentalis也迅速发布了回应公告。其中,Blackwell博士提到:“患者安全是首要任务,在临床试验中发生的任何死亡事件都是不幸的。公司正在与FDA密切合作,以尽快解决部分临床搁置问题。”
但是,如果抱着最坏的预期来看这件事,即最终Zentalis的三项研究经协调,仍被FDA认为风险大,应该停止。那么这就意味着,Zentalis管线中针对Azenosertib的单药治疗研究几乎将全军覆没。
目前,从Zentalis管线来看,除了上述三项已“危”的单药临床研究,目前Zentalis关于这款WEE1抑制剂仍有多项联合疗法在研。其中,包括与PARP抑制剂联用治疗PARP耐药的卵巢癌的MAMMOTH研究、与化疗联用治疗卵巢癌的研究及与吉西他滨联用治疗成人/儿童复发难治性骨肉瘤的研究等。
图片来源:Zentalis官网
值得注意的是,包括GSK和辉瑞都曾前后与Zentalis达成合作,就Azenosertib进行肿瘤相关研究,均为联合用药,不受此次事件影响。
另外,在今年5月发布的Q1财报中,Zentalis曾对Azenosertib的首个新药申请(NDA)作出预测。据披露,其有可能在2026年提交首个针对Azenosertib的新药上市申请,并透露这份申请将与“妇科恶性肿瘤”治疗有关。
02
Wee1赛道全景
如果Zentalis在其Q1公告中提到的消息确定能落实执行,那么就像这家公司公开展示在其官网的信息一样,Azenosertib将有望赛道第一。
图片来源:Zentalis官网
但是,如果各位读者对这个靶点有所了解的话,应该知道WEE1抑制剂赛道中最早被媒体关注到的,其实是阿斯利康。2013年9月,阿斯利康跟默沙东达成协议,阿斯利康将获得默沙东的一项已经进行到 IIa期临床Wee1抑制剂的全球权益。
图片来源:阿斯利康官网
目前,根据智慧芽数据库信息,布局Wee1赛道的企业并不多,进入II期临床阶段的管线除了Zentalis的Azenosertib,还有阿斯利康的Adavosertib,以及石家庄智康弘仁新药开发有限公司的SC-0191。
单看国内药企,除了智康弘仁,南京英派药业(I期),首药控股(I期),上海迪诺(临床前),上海医药集团(临床前)等也均有相关布局。
03
小结
从智慧芽数据库信息得知,在研的靶向Wee1的产品大部分均为小分子化药。现如今,在肿瘤领域已经出现CAR-T、基因疗法等更前沿上市产品时,用于肿瘤治疗的小分子化药热度似乎不如以前。
早前,因IRA法案中对生物制剂与小分子化药在保护期方面的区别对待,就有一部分声音表示,这将让企业考虑到成本问题而“抛弃”小分子项目转投生物制剂。
但其实在真实世界中小分子化药在肿瘤治疗中仍占据很大比例。其中原因直指患者端的支付能力。
就在近日,国际骨髓瘤基金会领导的改善癌症护理准入联盟(CIACC)的一场国会简报会就“我有保险,为什么却负担不起口服抗癌药物?”开展了相关讨论。
会议上,来自患者方的Tony Newberne分享道:“为了活下去,我支付了3000美元,但我知道我没有长期负担这笔钱的能力。”
此次会议在《癌症药物平价法案》基础上展开,该法案旨在确保口服和静脉化疗的更广泛的覆盖率。
参考资料:
1."I Have Insurance, Why Can't I Afford My Oral Anticancer Medications?" International Myeloma Foundation Highlights the Urgent Need for Cancer Drug Parity Act at Congressional Briefing
2.各公司官网
3.其他公开资料
封面图来源:Pixabay
股价暴涨477%,CD20 CAR-T再突破
>50%破发!2024年纳斯达克IPO的biotech公司依然面临挑战
MNC新药管线大比武!罗氏蝉联榜首,辉瑞第二,恒瑞首次上榜,排名第八
点击这里,发现价值信息!
ASCO会议免疫疗法财报临床2期快速通道
2022-11-07
·药时代
「谋划」未来,「实践」真知!「挑战」已至,「政策」先行!——第三届中国新药CMC高峰论坛精彩回顾!Wee1 激酶最早发现于裂殖酵母中,是丝氨酸/苏氨酸蛋白激酶家族的重要成员之一。它是一种高度保守的细胞周期调节蛋白,主要由3 个结构域组成,即N-端结构域、中心激酶结构域和1 个短的C-端结构域。N-端结构域是Wee1 激酶的激活结构域,也是抑制周期蛋白依赖性激酶1-周期蛋白B(CDK1-CyclinB)复合物去磷酸化的潜在位点,这些泛素化识别位点可以严格控制Wee1 的细胞内半衰期;C-端结构域主要包含催化段和活化段。Wee1 激酶细胞分裂间期S期能磷酸化CDK1的第15 位酪氨酸,进而抑制CDK1活性;在G2 期,DNA 复制完成后,Wee1 转而磷酸化组蛋白2B(H2B)的37 位酪氨酸,抑制转录共激活因子核蛋白(NPAT)和RNA 聚合酶Ⅱ的结合,并招募上游组蛋白1(Hist1)的分子伴侣HIR 组蛋白细胞周期调控缺陷同源物A(HIRA),下调H2B 表达,抑制S 期末期组蛋白转录,维系DNA-组蛋白的化学计量比。polo样蛋白激酶1(PLKl)磷酸化激活周期蛋白25 同源蛋白C(Cdc25C),Cdc25C 通过去磷酸化激活CDK1, 活化的CDK1 结合CyclinB 形成CDK1-CyclinB复合物,驱动细胞进入分裂期。然而,基因毒性应激、烷基化剂、辐射等刺激导致DNA 发生单链或双链断裂,DNA 损伤反应信号通路(DDR) 被激活,从而激活下游共济失调毛细血管扩张激酶(ATM) 信号通路或共济失调毛细血管扩张Rad3 相关激酶(ATR) 信号通路。Wee1 激酶通过2 条途径阻滞细胞有丝分裂:1)磷酸化组蛋白H2B 的37 位酪氨酸,抑制转录共激活因子NPAT和RNA聚合酶Ⅱ的结合,并招募了上游组蛋白伴侣HIRA,下调H2B 表达,产生S 期阻滞;2)抑制Cdc25C 从而抑制CDK1 去磷酸化或直接磷酸化CDK1 的第15 位酪氨酸,抑制CDK1活性,产生G2/M 阻滞,为修复受损DNA争取时间,维持染色质的完整性。合成致死(SL)是指2 个或多个非致死基因同时失活导致细胞死亡的现象。在正常细胞中,DNA 损伤还可通过抑癌基因p53介导的2种通路:ATM/ATR-P53-CDK4/CyclinD 和ATM/ATR-P53-CDK2/CyclinE。然而,在肿瘤细胞中,抑癌基因p53 突变率较高,超过50% 的肿瘤细胞中存在该基因的突变。p53 功能缺陷的肿瘤细胞G1/S 检查点失活,这使得DNA修复主要依赖于G2/M 检查点。Wee1抑制剂能够驱动细胞过早进入分裂期,从而产生SL 效应。理论上,抑制Wee1 激酶活性能够选择性地杀死p53 功能缺陷肿瘤细胞,而不影响正常细胞,相关研究表明,Wee1 激酶在肝癌、肺癌、结直肠癌、胃癌、乳腺癌、卵巢癌、宫颈癌、鳞状细胞癌、恶性黑色素瘤、胶质母细胞瘤、桥内弥漫性脑胶质瘤中过表达,且在卵巢癌、黑色素瘤和胶质母细胞瘤中的高表达与不良预后相关,是一种理想的肿瘤治疗途径。01最新研究进展1、据不完全统计,目前在研的Wee1药物十余种,其中包括Wee1激酶抑制剂和PROTAC。其中Wee1激酶抑制剂按结构类型可分为嘧啶并吡啶酮类、嘧啶并吡唑酮类、咔唑并吡咯二酮类及其他类。2、在研究企业上,其中AI制药公司Schrödinger,以及国外企业阿斯利康、辉瑞、Debiopharm等均在该领域布局;中国公司英派药业、首药控股、迪诺医药、智康弘仁也均在该领域进行布局。3、将在研的Wee1药物统计如下:02重点产品介绍1、ZN-c3ZN-c3是一款口服的Wee1抑制剂,目前由Zentalis和辉瑞共同开发,旨在诱导癌细胞中的合成致死,目前正在开发用于晚期实体瘤。此前,FDA已授予口服ZN-c3快速通道资格,用于治疗复发或持久性子宫浆液性癌。2022年10月,Zentalis宣布,计划启动ZN-c3 与encorafenib 和西妥昔单抗(BEACON方案)联合治疗 BRAF V600E 突变的转移性结直肠癌(mCRC) 患者的1/2 期剂量递增研究。2022年4月,Zentalis在AACR大会上公布了ZN-c3与化疗联用,治疗对含铂化疗耐药,或难治性卵巢癌患者的1b期临床试验数据,结果显示:在43名可以评估疗效的患者中,组合疗法达到30.2%的客观缓解率(ORR)和86.0%的疾病控制率(DCR)。2、adavosertibAdavosertib 是首个进入临床的Wee1 小分子抑制剂,2012 年在欧盟获批孤儿药资格,用于治疗卵巢癌。2013 年,默沙东将该产品的全球开发和营销授权给阿斯利康。目前主要开发用于治疗卵巢癌和胰腺癌。在铂耐药的上皮性卵巢癌、输卵管癌或原发性腹膜癌患者中,评估了adavosertib 联合卡铂、吉西他滨治疗的有效性和安全性。显示与吉西他滨联用组ORR和中位PFS 分别为31.9% 和5.5 个月;与卡铂联用组的疗效最好,其ORR 为66.7%,中位PFS 为12 个月。除此以外,adavosertib在II期卵巢癌、胰腺癌试验中,均展示出良好的治疗前景。3、Debio 0123Debio 0123是Debiopharm开发的一种口服小分子Wee1激酶抑制剂。体外实验表明,Debio 0123能够通过抑制CDK1磷酸化,剂量依赖性地诱导DNA损伤,从而促进肿瘤细胞凋亡;动物模型显示,在无胸腺裸鼠的A427皮下异种移植瘤模型中,30mg·kg-1剂量连续口服给药28d,肿瘤明显消退。目前,该药物与卡铂联合治疗复发或难治性局部晚期或转移性实体瘤正在进行中,预计于2022年12月完成。4、IMP7068IMP7068是英派药业自主研发的Wee1抑制剂,动物实验显示,它在动物体内的半衰期及药物暴露量均优于同类在研药物,有望成为best-in-class的药物。2022年9月,公司在2022 ESMO上公布IMP7068在晚期实体瘤患者中的安全性、药代动力学和药效学特征的初步数据,显示在8个爬坡队列中纳入的32例患者,中位随访时间为212天,其中一例患者(子宫浆液性癌)最佳疗效评估达到部分缓解。与研究药物相关的不良事件发生率为65.6%,大多数为1-2级不良事件。说明IMP7068具有良好的耐受性和对WEE1的抑制作用,其药代动力学和药效学特征与剂量水平一致。5、SY-4835SY-4835是由首药控股开发的选择性WEE1抑制剂,临床前研究结果表明,SY-4835对多种肿瘤均有显著抑制活性,抗癌谱广泛。目前,该药正在进行I期临床试验,其潜在开发适应症包括胰腺癌、卵巢癌、乳腺癌等多种实体瘤。版权声明/免责声明本文为授权转载内容,版权归拥有者。仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。欢迎朋友们批评指正!衷心感谢!文中图片、视频为授权正版作品,或来自微信公共图片库,或取自公司官网/网络根据CC0协议使用,版权归拥有者。任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!中国制药江湖冬天里的一把火!请欣赏图片直播!已被欣赏8万+点击这里,欣赏中国制药江湖「冬天里的一把火」!
免疫疗法快速通道Best in Class抗体小分子药物
2022-09-21
·研发客
特发性肺纤维化(IPF)是一种慢性纤维化间质性肺炎,发病机制不明,且预后不良。目前只有两款治疗药物—吡非尼酮和尼达尼布上市,他们主要延缓肺功能下降,延缓IPF进程。尽管疗效有限且耐受性差,这两款药物在2021年仍达成11亿和25亿美元的销售额。鉴于IPF的发病机制不明确、患者人数少,在中国被纳入罕见病目录,限制了其药物开发和临床研究。但对IPF的研究从未停止,近年来相继有创新靶点药物被开发出来并进入临床研究阶段。根据研发客研究院统计,国内共有21款产品开展IPF临床研究,药物靶点较为分散且进展缓慢,进入临床III期仅有罗氏的RG6354和FibroGen公司的pamrevlumab。国内企业自主研发进入II期临床的产品有泰德制药的TDI01、众生药业的ZSP1603、泽璟制药的杰克替尼、天方药业的YPS345,以及两款二代吡非尼酮,分别是爱科百发的AK3280和东阳光药的伊非尼酮。两款进入III期临床pamrevlumab(FG3019)是FibroGen公司开发的一种人源性CTGF单克隆抗体,正在开展治疗IPF的III期临床研究,是目前唯一证明在IPF患者中有临床疗效的抗体药物。一项II期随机、双盲、安慰剂对照试验(PRAISE)中,在第48周时,主要疗效终点—用力肺活量(FVC)百分比占预计值的下降比例,pamrevlumab组相比安慰剂组降低了60.3%,pamrevlumab组疾病进展的患者比例低于安慰剂组,耐受性良好,安全性与安慰剂相似。4月,FibroGen宣布完成III期临床ZEPHYRUS-1的首例患者给药,预计在2023年完成试验。抗结缔组织生长因子(CTGF)是一种多效生长因子,参与上皮-间充质转化、胶原沉积和纤维化细胞过程,通常低水平表达,但在几乎所有纤维化病症中显著富集。国内此领域在研的产品很少,恒瑞医药正在开展I期临床的SHR-1906,虽未公开靶点,但根据恒瑞医药的专利信息推测,其有可能是CTGF抗体。国外Pieris Pharmaceuticals的PRS-220在I期临床中。另一进展至III期临床的产品是罗氏的RG6354。RG6354(曾用名PRM-151)是罗氏在2019年通过收购Promedior公司获得,是内源性人类先天免疫蛋白PTX-2的重组类似物,已被FDA授予治疗IPF的突破性疗法认定。研究表示,PTX-2(pentraxin-2)可有效抑制由TGFβ1驱动的所有促纤维化不良病变,给包含IPF在内所有纤维化疾病的治疗提供了新思路。Promedior公开的II期数据显示,117名IPF患者接受28周治疗后,RG6354组的FVC预测值百分比变化为-2.5%,减缓肺功能下降明显优于安慰剂组的-4.8%,此外患者依从性达到97%。在失败中探索IPF本身疾病机制难以明确,且患者人数不多,2020年全球约有160万人受影响,已被收录于国家《第一批罕见病目录》中,这给IPF治疗药物的研发增加一定难度,相继有企业因各类原因终止产品开发。例如在抗炎途径IL-13和IL-4拮抗剂领域,礼来在2013年启动IL-13单抗lebrikizumab的II期临床后未见有进一步结果公布,赛诺菲于2017年终止了IL4/IL13双特异性抗体romilkimab治疗IPF的研究。被放弃的项目中,ATX抑制剂ziritaxestat(GLPG1690)是唯一一款进入III期研究宣告失败的产品。2021年2月,Galapagos与吉利德联合宣布,从获利-风险考虑终止ziritaxestat治疗IPF的III期研究ISABELA,并停止产品所有临床试验。同领域的其他药物仍在积极开展临床研究。中国公司恒诺康医药研发的HNC664已经完成治疗IPF的Ia期临床研究,并布局了二代ATX抑制剂HNC1058,产品未申报临床。国外方面,Bridge Biotherapeutics将BBT-877推进至II期临床。据研发客研究院统计,ATX-LPA通路是IPF药物开发针对最多的靶点。有研究发现在博来霉素肺纤维化模型小鼠及IPF患者中均发现溶血磷脂酸(LPA)水平升高,自分泌运动因子(autotaxin,ATX)是一种生成血液中LPA的关键细胞外酶。溶血磷脂酸受体1(LPA1)拮抗剂开发中,百时美施贵宝的BMS-986278在中国启动了II期临床,另外,公司还有一款同类产品BMS-986337处于临床I期阶段,一款BMS-986020在临床前。ziritaxestat临床失败给ATX-LPA通路研究增加了不确定性。另一方面,整合素抑制剂在IPF领域的探索也喜忧参半。2022年7月,Pliant Therapeutics宣布PLN-74089治疗IPF的IIa期临床研究达到主要疗效终点,PLN-74809剂量依赖性的显著延缓FVC下降,且具备良好的耐受性。PLN-74809是αvβ6/avβ1双重整合素抑制剂,通过抑制纤维化组织中的关键介质TGFβ通路,发挥治疗纤维化的作用。渤健开发的αvβ6整合素拮抗剂BG00011在2019年9月因安全性问题终止临床。更多在研创新产品特发性肺纤维化的未满足需求吸引众多国内企业布局,这些在研产品靶点分散,产品大部分处于早期临床阶段。其中进入到临床II期阶段是泽璟制药的杰克替尼、勃林格殷格翰的BI 1015550、众生药业的ZSP1603、天方药业的YPS345。杰克替尼是一款口服JAK抑制剂,泽璟制药在2020年7月启动了治疗IPF的II期临床研究。勃林格殷格翰开发的PDE4B(磷酸二酯酶4B)抑制剂BI 1015550,于近期在国内递交了治疗IPF的临床申请。公司5月于NEJM杂志公布的临床数据显示,BI 1015550能够有效减缓IPF患者的肺功能衰退,在减缓IPF患者肺功能恶化方面优于安慰剂的概率大于98%。在血小板衍生生长因子(platelet-derived growth factor,PDGF)抗体领域,国内公司众生药业开发的ZSP1603正处于II期临床试验中,IPF适应症上,是国内同靶点第一个获批临床的产品。YPS345是天方药业与中国科学院生物物理研究共同开发的1类新药,正在开展治疗胸部肿瘤放疗引起的肺炎及肺纤维化的II期临床试验,产品靶点未公开。据研发客研究院统计,10个处于I期临床阶段的药物中,有很多公司未公开其产品靶点,例如人福医药的HW021199、海思科的FTP-198、智康弘仁的SC-1011以及普沐生物的PMG1015。值得一提的是AI制药企业英矽智能的ISM001-055,是公司首款由AI发现设计、治疗IPF的产品,靶点未公开。据英矽智能公布信息,ISM001-055靶向公司通过PandaOmics平台发掘出的新颖纤维化靶点Target X,该靶点与多种纤维化疾病相关。7月,英矽智能宣布ISM001-055完成了I期临床的首例患者给药。TDI01是一款高选择性ROCK2抑制剂,由中国生物制药附属公司泰德制药自主研发,目前正在开展治疗IPF的I期临床试验。这款药物还在2021年2月成功出海,Graviton Bioscience公司获得其在大中华以外地区共同开发和商业化权益,泰德制药获得的首付款及研发、销售里程碑付款最高可达5.175亿美元。安立玺荣生物开发的CSF-1R拮抗剂EI-1071在2022年8月被FDA授予治疗IPF的孤儿药资格认定,处在I期临床研究中。同类产品中,国内还有宝船生物的BC006在临床前阶段。复星医药子公司复星弘创自主研发的新型选择性肌醇酶(IRE1)抑制剂ORIN1001,已在美国完成治疗IPF的Ib期临床,国内已获得IND批准。间充质干细胞(MSCs)也被开发用来治疗IPF,国内公司中,莱馥医疗的人脐带间充质干细胞、生创精准的SC01009(宫血间充质干细胞)都开展了治疗IPF的I期临床研究。除了上述产品,国内还有部分公司在已上市的吡非尼酮基础上开发二代产品。临床应用结果显示吡非尼酮的耐受性差、疗效有限,约有一半患者使用吡非尼酮后,选择停止治疗、减少剂量或改用其他治疗方式。爱科百发和东阳光药开发的二代吡非尼酮,能一定程度上延缓一代吡非尼酮的耐受。东阳光药业的伊非尼酮已步入临床II期研究,爱科百发在2022年5月宣布启动AK3280治疗IPF的II期临床。总第1712期访问研发客网站可浏览更多文章www.PharmaDJ.com
孤儿药并购免疫疗法突破性疗法
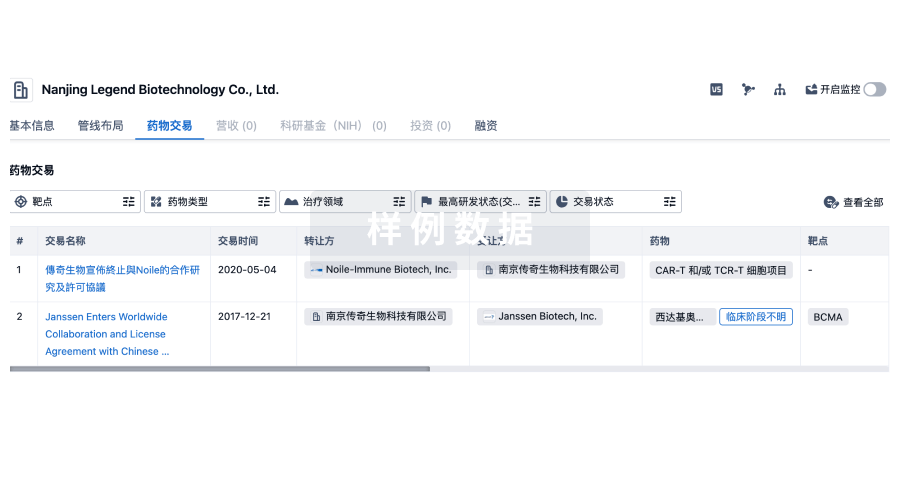
100 项与 无锡智康弘仁新药开发有限公司 相关的药物交易
登录后查看更多信息
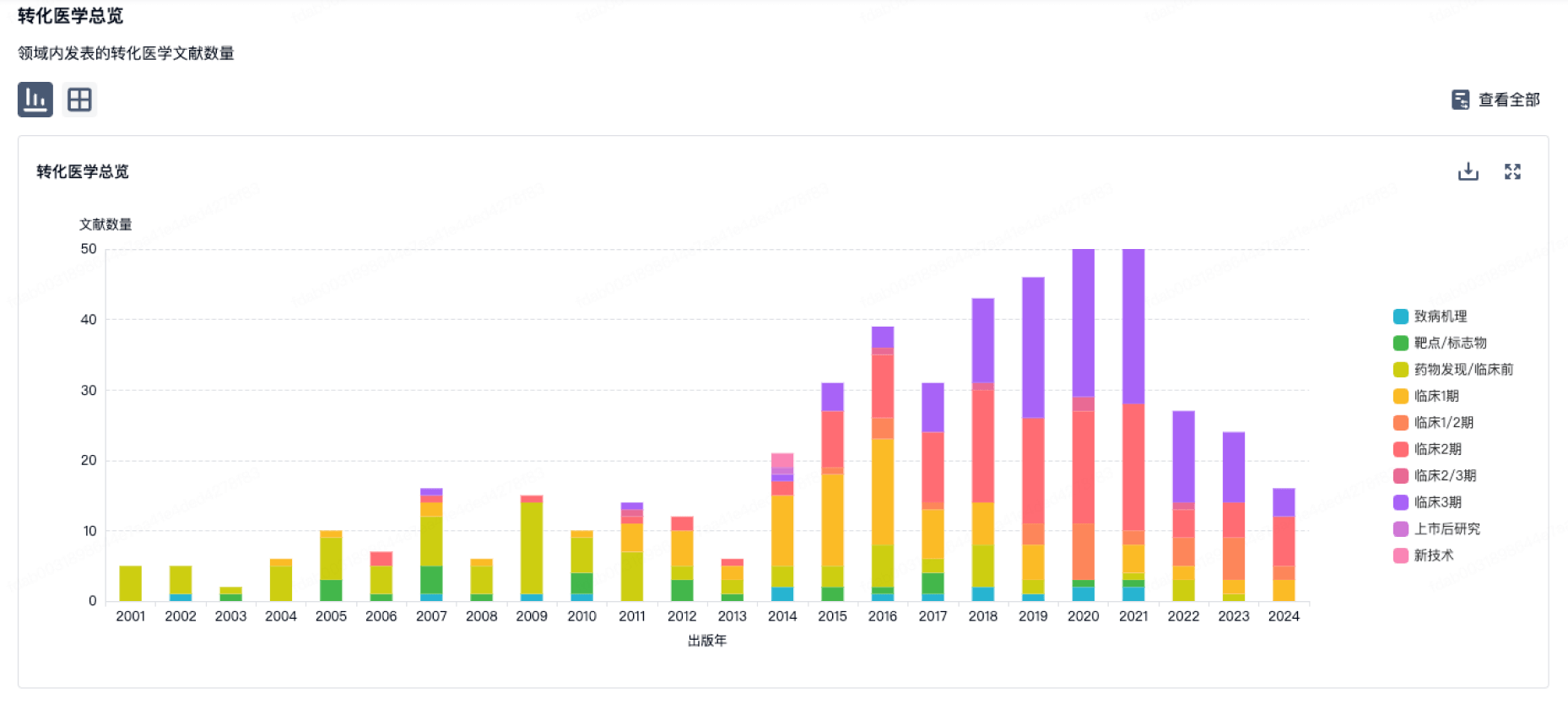
100 项与 无锡智康弘仁新药开发有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年11月04日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
临床2期
1
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用