预约演示
更新于:2025-05-07

Istituto Giannina Gaslini
更新于:2025-05-07
概览
标签
肿瘤
其他疾病
耳鼻咽喉疾病
ADC
合成多肽
脂质体药物
疾病领域得分
一眼洞穿机构专注的疾病领域

技术平台
公司药物应用最多的技术
靶点
公司最常开发的靶点
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
NCT06625112
A Multicentric European Study to Promote Clinical Trial Readiness for STXBP1-related Disorders
NCT06515080
Physiological Monitoring in the OR; Predicting Outcomes Using Infra-red SEnsors, a Feasibility Study.

NCT06618937
Toward Personalized Medicine to Guide Drug Withdrawal in Children with Juvenile Idiopathic Arthritis in Clinical Remission: a Randomized Clinical Trial Comparing Early Versus Late Drug Withdrawal Combining Imaging and Multi-Omics
100 项与 Istituto Giannina Gaslini 相关的临床结果
登录后查看更多信息
登录后查看更多信息
2025-02-27Journal of Medicinal Chemistry
Target Identification with Live-Cell Photoaffinity Labeling and Mechanism of Action Elucidation of ARN23765, a Highly Potent CFTR Corrector
Article
作者: Pedemonte, Nicoletta ; Laselva, Onofrio ; Ocello, Riccardo ; Allegretta, Caterina ; Saccoliti, Francesco ; Falchi, Federico ; Romeo, Elisa ; Andonaia, Angela ; Pastorino, Cristina ; Bandiera, Tiziano ; Bertozzi, Fabio
2025-02-01Neuroradiology
Retinoblastoma and beyond: pediatric orbital mass lesions
Review
作者: Rossi, Andrea ; Clarke, Rebekah ; Galluzzi, Paolo ; Dodig, Doris ; Brumini, Ivan ; Rumboldt, Zoran ; Singh, Sumit
2024-12-01Journal of Neurology
Correction to: CDKL5 deficiency-related neurodevelopmental disorders: a multi-center cohort study in Italy
作者: Savasta, Salvatore ; Striano, Pasquale ; Matricardi, Sara ; Operto, Francesca Felicia ; Fattorusso, Antonella ; Verrotti, Alberto ; Ferretti, Alessandro ; Mastrangelo, Mario ; Salpietro, Vincenzo ; Dell'Isola, Giovanni Battista ; Carotenuto, Marco ; Scorrano, Giovanna ; Fruttini, Daniela ; Parisi, Pasquale ; Dell’Isola, Giovanni Battista ; Pisani, Francesco ; Prontera, Paolo ; Pruna, Dario ; Di Cara, Giuseppe ; Spalice, Alberto ; Pavone, Piero ; Elia, Maurizio ; Spezia, Elisabetta ; Cordelli, Duccio Maria
100 项与 Istituto Giannina Gaslini 相关的药物交易
登录后查看更多信息
100 项与 Istituto Giannina Gaslini 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年07月22日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
临床前
2
1
临床1期
登录后查看更多信息
当前项目
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
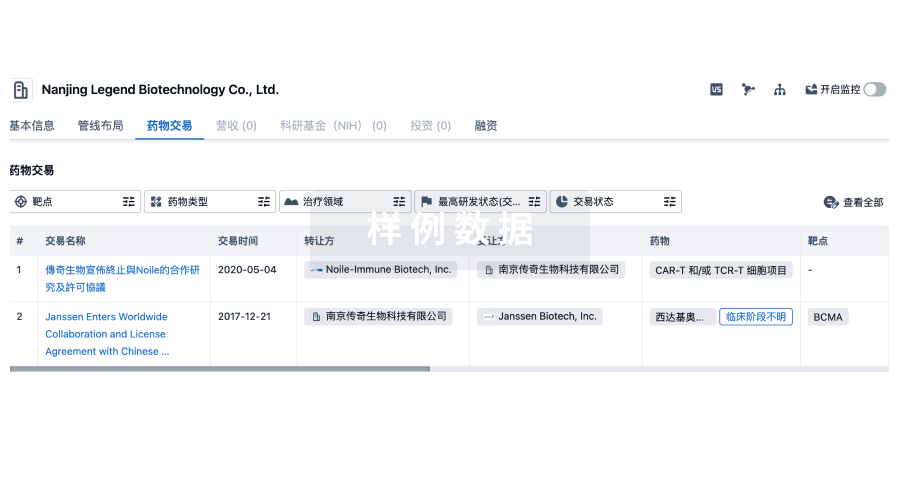
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用