预约演示
更新于:2025-07-09
Nivolumab Biosimilar(Luye Pharma)
纳武利尤单抗生物类似药(绿叶制药)
更新于:2025-07-09
概要
基本信息
药物类型 生物类似药、单克隆抗体 |
别名 Nivolumab Biosimilar (Boan Biological)、纳武利尤单抗生物类似药 (博安生物)、纳武利尤单抗生物类似药(Luye Pharma Group Ltd.) + [5] |
靶点 |
作用方式 抑制剂 |
作用机制 PD-1抑制剂(细胞程序性死亡-1抑制剂) |
在研适应症 |
非在研适应症- |
原研机构 |
非在研机构- |
权益机构- |
最高研发阶段临床3期 |
首次获批日期- |
最高研发阶段(中国)临床3期 |
特殊审评- |
登录后查看时间轴
结构/序列
Sequence Code 93681H

当前序列信息引自: *****
Sequence Code 93684L
当前序列信息引自: *****
关联
4
项与 纳武利尤单抗生物类似药(绿叶制药) 相关的临床试验CTR20232393
比较LY01015与欧狄沃®(纳武利尤单抗注射液)分别联合氟尿嘧啶和顺铂用于治疗晚期或转移性食管鳞癌患者的有效性和安全性随机、双盲、多中心III期临床研究
NCT06022861
A Randomized, Double-blind, Multicenter, Phase 3 Study to Compare the Efficacy and Safety of LY01015 and Opdivo®(Nivolumab Injection)Combined Respectively With Fluorouracil Plus Cisplatin in Participants With Advanced or Metastatic Esophageal Squamous Cell Carcinoma.
ChiCTR2200064771
A randomized, double-blind, single-dose, parallel-controlled phase I clinical study comparing the pharmacokinetic profile, safety, tolerability and immunogenicity of LY01015 and Ondivololol? (nabumab injection) in healthy Chinese male subjects
100 项与 纳武利尤单抗生物类似药(绿叶制药) 相关的临床结果
登录后查看更多信息
100 项与 纳武利尤单抗生物类似药(绿叶制药) 相关的转化医学
登录后查看更多信息
100 项与 纳武利尤单抗生物类似药(绿叶制药) 相关的专利(医药)
登录后查看更多信息
1
项与 纳武利尤单抗生物类似药(绿叶制药) 相关的文献(医药)2024-11-01BIODRUGS
Pharmacokinetics, Safety, and Immunogenicity of a Biosimilar of Nivolumab (LY01015): A Randomized, Double-Blind, Parallel-Controlled Phase I Clinical Trial in Healthy Chinese Male Subjects
Article
作者: Dou, Changlin ; Song, Hongtao ; Deng, Kunhong ; Yang, Guoping ; Hu, Baihui ; Huang, Jie ; Qi, Fan ; Wang, Wei ; Zhang, Shengnan ; Liu, Li ; Xie, Jinlian ; Zhou, Ming ; Ye, Ling ; Zhao, Yanyan ; Cui, Chang ; Wu, Qian ; Li, Xiaojing
BACKGROUND:
Nivolumab (Opdivo®) is the first anti-PD-1 antibody approved in the world. LY01015 is a potential biosimilar of nivolumab.
OBJECTIVES:
This phase I study aimed to establish the pharmacokinetic equivalence between LY01015 and the original investigational nivolumab (Opdivo®) in healthy Chinese male subjects. Additionally, safety and immunogenicity were assessed.
PATIENTS AND METHODS:
A randomized, double-blind, parallel-controlled, phase I trial was conducted with 176 healthy male adults receiving a single intravenous infusion of LY01015 or nivolumab at 0.3 mg/kg. Pharmacokinetics, safety, and immunogenicity were evaluated over a 99-day period. The primary pharmacokinetics endpoint was AUC0-∞, and the secondary pharmacokinetic endpoints included AUC0-t and Cmax. Pharmacokinetic bioequivalence was confirmed using standard equivalence margins of 80.00-125.00%.
RESULTS:
This study is the first to report on the pharmacokinetics, safety, and immunogenicity of Opdivo® in healthy individuals. The pharmacokinetics profiles of LY01015 and Opdivo® were found to be comparable. The geometric mean ratios (90% confidence intervals) for the AUC0-∞, AUC0-t, and Cmax of LY01015 to Opdivo® were 94.49% (90.29-98.88%), 94.92% (88.73-101.54%), and 96.55% (93.32-99.90%), respectively, falling within the conventional bioequivalence criteria of 80.00-125.00%. The safety and immunogenicity were also comparable between the two groups.
CONCLUSIONS:
LY01015 demonstrated highly similar pharmacokinetics to nivolumab in healthy Chinese male subjects. Both drugs exhibited comparable safety and immunogenicity profiles.
TRIAL REGISTRATION:
This trial is registered at the Chinese Clinical Trial Registry website ( https://www.chictr.org.cn/ #ChiCTR2200064771).
30
项与 纳武利尤单抗生物类似药(绿叶制药) 相关的新闻(医药)2025-06-18
·摩熵医药
临床申请siRNA信使RNA
2025-04-29
·博安生物
AACR会议临床结果临床1期
2025-03-27
·博安生物
财报抗体药物偶联物临床1期AACR会议生物类似药
100 项与 纳武利尤单抗生物类似药(绿叶制药) 相关的药物交易
登录后查看更多信息
外链
| KEGG | Wiki | ATC | Drug Bank |
|---|---|---|---|
| - | - | - |
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 食管鳞状细胞癌 | 临床3期 | 中国 | 2023-10-12 | |
| 转移性食管鳞状细胞癌 | 临床3期 | 中国 | 2023-10-12 | |
| 不能切除的食管鳞状细胞癌 | 临床3期 | 中国 | 2023-10-12 | |
| 食管腺癌 | 临床3期 | 中国 | - | |
| 结直肠癌 | 临床3期 | 中国 | - | |
| 胃食管交界处癌 | 临床3期 | - | - | |
| 肝细胞癌 | 临床3期 | 中国 | - | |
| 霍奇金淋巴瘤 | 临床3期 | 中国 | - | |
| 恶性胸膜间皮瘤 | 临床3期 | 中国 | - | |
| 黑色素瘤 | 临床3期 | - | - |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
临床1期 | - | 176 | 顧願艱壓窪鏇窪蓋觸鏇(鑰觸糧淵願醖鏇鬱顧獵): Ratio = 94.49% (90% CI, 90.29 ~ 98.88) 更多 | 积极 | 2024-09-24 | ||
Opdivo |

登录后查看更多信息
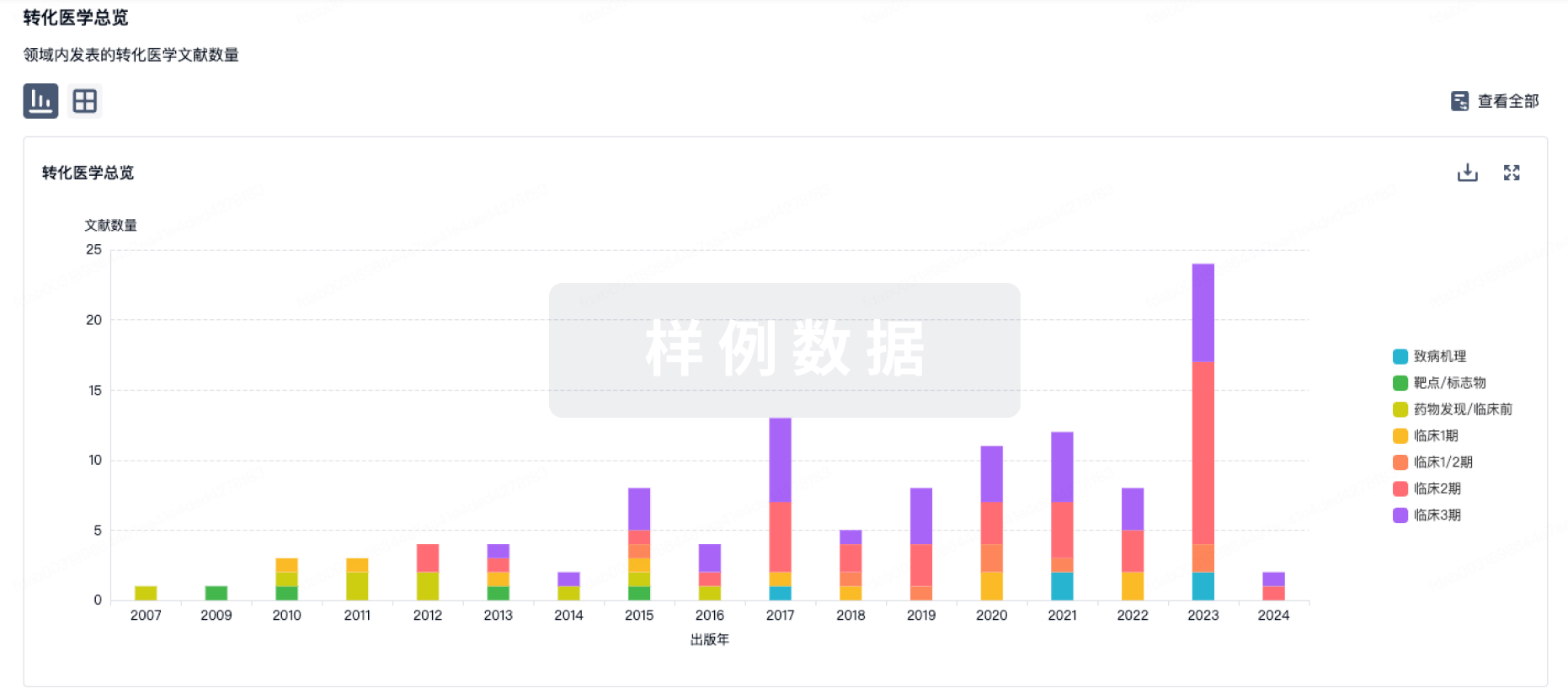
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用