预约演示
更新于:2025-06-14
ATP-binding transporter gene therapy(Oxford Biomedica Plc)
更新于:2025-06-14
概要
基本信息
在研机构 |
最高研发阶段临床2期 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |
登录后查看时间轴
关联
2
项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的临床试验NCT01736592
An Open Label Study to Determine the Long Term Safety, Tolerability and Biological Activity of SAR422459 in Patients With Stargardt's Macular Degeneration
Primary Objective:
To evaluate the long-term safety and tolerability of SAR422459 in patients with Stargardt's Macular Degeneration.
Secondary Objective:
To assess:
* Safety
* Biological activity
To evaluate the long-term safety and tolerability of SAR422459 in patients with Stargardt's Macular Degeneration.
Secondary Objective:
To assess:
* Safety
* Biological activity
开始日期2012-12-14 |
申办/合作机构 |
NCT01367444
A Phase I/IIA Dose Escalation Safety Study of Subretinally Injected SAR422459, Administered to Patients With Stargardt's Macular Degeneration
Primary Objective:
To assess the safety and tolerability of ascending doses of SAR422459 in participants with Stargardt's Macular Degeneration (SMD).
Secondary Objective:
To evaluate for possible biological activity of SAR422459.
To assess the safety and tolerability of ascending doses of SAR422459 in participants with Stargardt's Macular Degeneration (SMD).
Secondary Objective:
To evaluate for possible biological activity of SAR422459.
开始日期2011-06-08 |
申办/合作机构 |
100 项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的临床结果
登录后查看更多信息
100 项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的转化医学
登录后查看更多信息
100 项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的专利(医药)
登录后查看更多信息
3
项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的文献(医药)2017-08-01·Heliyon
Strategies for enzyme saving during saccharification of pretreated lignocellulo-starch biomass: effect of enzyme dosage and detoxification chemicals
Article
作者: Padmaja, G ; Mithra, M G
Two strategies leading to enzyme saving during saccharification of pretreated lignocellulo-starch biomass (LCSB) was investigated which included reducing enzyme dosage by varying their levels in enzyme cocktails and enhancing the fermentable sugar yield in enzyme-reduced systems using detoxification chemicals. Time course release of reducing sugars (RS) during 24-120 h was significantly higher when an enzyme cocktail containing full dose of cellulase (16 FPU/g cellulose) along with half dose each of xylanase (1.5 mg protein/g hemicelluloses) and Stargen (12.5 μl/g biomass) was used to saccharify conventional dilute sulphuric acid (DSA) pretreated biomass compared to a parallel system where only one-fourth the dose of the latter two enzymes was used. The reduction in RS content in the 120 h saccharified mash to the extent of 3-4 g/L compared to the system saccharified with full complement of the three enzymes could be overcome considerably by supplementing the system (half dose of two enzymes) with detoxification chemical mix incorporating Tween 20, PEG 4000 and sodium borohydride. Microwave (MW)-assisted DSA pretreated biomass on saccharification with enzyme cocktail having full dose of cellulase and half dose of Stargen along with detoxification chemicals gave significantly higher RS yield than DSA pretreated system saccharified using three enzymes. The study showed that xylanase could be eliminated during saccharification of MW-assisted DSA pretreated biomass without affecting RS yield when detoxification chemicals were also supplemented. The Saccharification Efficiency and Overall Conversion Efficiency were also high for the MW-assisted DSA pretreated biomass. Since whole slurry saccharifcation of pretreated biomass is essential to conserve fermentable sugars in LCSB saccharification, detoxification of soluble inhibitors is equally important as channelling out of insoluble lignin remaining in the residue. As one of the major factors contributing to the cost of ethanol production from LCSB is the cost of enzymes, appropriate modification of enzyme cocktail based on the composition of the pretreated biomass coupled with effective detoxification of the slurry would be a promising approach towards cost reduction.
2016-10-01·Translational vision science & technology3区 · 医学
Test–Retest Variability of Functional and Structural Parameters in Patients with Stargardt Disease Participating in the SAR422459 Gene Therapy Trial
3区 · 医学
ArticleOA
作者: Yang, Paul ; Parker, Maria A. ; Steinkamp, Peter N. ; Erker, Laura R. ; Schlechter, Catherine L. ; Pennesi, Mark E. ; Sahel, Jose ; Dhaenens, Claire-Marie ; Choi, Dongseok ; Audo, Isabelle ; Weleber, Richard G. ; Mohand-Said, Saddek ; Wilson, David J. ; Chegarnov, Elvira N.
PURPOSE:
The goal of this analysis was to determine the test-retest variability of functional and structural measures from a cohort of patients with advanced forms of Stargardt Disease (STGD) participating in the SAR422459 (NCT01367444) gene therapy clinical trial.
METHODS:
Twenty-two participants, aged 24 to 66, diagnosed with advanced forms of STGD, with at least one pathogenic ABCA4 mutation on each chromosome participating in the SAR422459 (NCT01367444) gene therapy clinical trial, were screened over three visits within 3 weeks or less. Functional visual evaluations included: best-corrected visual acuity (BCVA) Early Treatment Diabetic Retinopathy Study (ETDRS) letter score, semiautomated kinetic perimetry (SKP) using isopters I4e, III4e, and V4e, hill of vision (HOV) calculated from static visual fields (SVF) by using a 184n point centrally condensed grid with the stimulus size V test target. Retinal structural changes such as central macular thickness and macular volume were assessed by spectral-domain optical coherence tomography (SD-OCT). Repeatability coefficients (RC) and 95% confidential intervals (CI) were calculated for each parameter using a hierarchical mixed-effects model and bootstrapping.
RESULTS:
Criteria for statistically significant changes for various parameters were found to be the following: BCVA letter score (8 letters), SKP isopters I4e, III4e, and V4e (3478.85; 2488.02 and 2622.46 deg2, respectively), SVF full volume HOV (VTOT, 14.62 dB-sr), central macular thickness, and macular volume (4.27 μm and 0.15 mm3, respectively).
CONCLUSIONS:
This analysis provides important information necessary to determine if significant changes are occurring in structural and functional assessments commonly used to measure disease progression in this cohort of patients with STGD. Moreover, this information is useful for future trials assessing safety and efficacy of treatments in STGD.
TRANSLATIONAL RELEVANCE:
Determination of variability of functional and structural measures in participants with advanced stages of the STGD is necessary to assess efficacy and safety in treatment trials involving STGD patients.
2013-06-12·Investigative ophthalmology & visual science2区 · 医学
Transduction of Photoreceptors With Equine Infectious Anemia Virus Lentiviral Vectors: Safety and Biodistribution of StarGen for Stargardt Disease
2区 · 医学
ArticleOA
作者: Vezina, Mark ; Kelleher, Michelle ; Iqball, Sharifah ; Ellis, Scott ; Naylor, Stuart ; Gouras, Peter ; Prefontaine, Annick ; Hurst, Felicity ; Mitrophanous, Kyriacos A. ; Loader, Julie ; Allikmets, Rando ; de Belin, Jackie ; Ferrige, Georgina ; Wong, Paul ; Bergstrom, Christopher ; Carlucci, Marie ; Miskin, James ; Aaberg, Thomas ; Kong, Jian ; Angell-Manning, Diana ; Binley, Katie ; Widdowson, Peter ; Bussieres, Martin ; Fernandes, Alcides ; Yan, Jiong
PURPOSE:
StarGen is an equine infectious anemia virus (EIAV)-based lentiviral vector that expresses the photoreceptor-specific adenosine triphosphate (ATP)-binding cassette transporter (ABCA4) protein that is mutated in Stargardt disease (STGD1), a juvenile macular dystrophy. EIAV vectors are able to efficiently transduce rod and cone photoreceptors in addition to retinal pigment epithelium in the adult macaque and rabbit retina following subretinal delivery. The safety and biodistribution of StarGen following subretinal delivery in macaques and rabbits was assessed.
METHODS:
Regular ophthalmic examinations, IOP measurements, ERG responses, and histopathology were carried out in both species to compare control and vector-treated eyes. Tissue and fluid samples were obtained to evaluate the persistence, biodistribution, and shedding of the vector following subretinal delivery.
RESULTS:
Ophthalmic examinations revealed a slightly higher level of inflammation in StarGen compared with control treated eyes in both species. However, inflammation was transient and no overt toxicity was observed in StarGen treated eyes and there were no abnormal clinical findings. There was no StarGen-associated rise in IOP or abnormal ERG response in either rabbits or macaques. Histopathologic examination of the eyes did not reveal any detrimental changes resulting from subretinal administration of StarGen. Although antibodies to StarGen vector components were detected in rabbit but not macaque serum, this immunologic response did not result in any long-term toxicity. Biodistribution analysis demonstrated that the StarGen vector was restricted to the ocular compartment.
CONCLUSIONS:
In summary, these studies demonstrate StarGen to be well tolerated and localized following subretinal administration.
1
项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的新闻(医药)2021-04-15
Oxford Biomedica plc
Preliminary results for the year ended 31 December 2020
Saving Lives
Oxford, UK – 15 April 2021: Oxford Biomedica plc (LSE: OXB), (“OXB” or “the Group”), a leading cell and gene therapy group, today announces its preliminary results for the year ended 31 December 2020.
John Dawson, Chief Executive Officer of Oxford Biomedica, said:
“I am truly proud of the Group’s achievements over the period. We not only secured major new partnerships, brought the Oxbox manufacturing facility online in record time and responded to the challenges of the pandemic, but the team has also been able to rapidly work with AstraZeneca to provide a vaccine solution for COVID-19. This is a true testament to the world-class calibre and dedication of our staff in the year that the Group also gained entry to the FTSE250. Looking to the future, with the continued tide of growth in cell and gene therapy, coupled with the Group’s leadership position in the lentiviral vector field, we are well positioned to advance both our own proprietary pipeline and that of our current and future partners’ programmes. I would like to thank all of Oxford Biomedica’s employees for their hard work throughout 2020 and our shareholders and partners for their continued support, and I look forward to a successful 2021.”
FINANCIAL HIGHLIGHTS
Total revenues increased by 37% to £87.7 million (2019: Revenue of £64.1 million)
Bioprocessing and commercial development revenues increased by 45% to £68.5 million (2019: £47.3 million) with double digit growth across both activities, driven by new customers AstraZeneca, Beam Therapeutics and Juno/BMS
Revenues from licences, milestones & royalties increased to £19.2 million (2019: £16.8 million) due to the recognition of a £7.8 million ($10 million) licence fee from Juno/BMS as well as other licence fees, milestones and royalties from customers
Operating expenses 1 increased by less than revenues, growing by 23% in the year to £51.7 million (2019: £41.9 million) aided by the move to the lower cost bioreactor manufacturing process
increased by less than revenues, growing by 23% in the year to £51.7 million (2019: £41.9 million) aided by the move to the lower cost bioreactor manufacturing process Operating EBITDA 2 profit of £7.3 million (2019: £5.2 million loss), marginally above guided range
profit of £7.3 million (2019: £5.2 million loss), marginally above guided range Operating loss incurred of £5.7 million (2019: £14.5 million loss)
The platform segment generated an operating profit of £2.0 million ( 2019: 20.2 million loss) whilst the Product segment made a loss of £7.7 million ( 2019: £5.7 million profit)
Capital expenditure decreased to £13.4 million (2019: £25.8 million) mainly reflecting the Windrush Court laboratory conversion and equipment purchases and leasehold improvements at Oxbox
Cash of £46.7 million at 31 December 2020 (2019: £16.2 million) and £65.9 million at 31 March 2021
Cash used in operations of £3.9 million in 2020 (2019: £6.6 million) decreased as a result of the increased revenues as explained above, offset by further operational investments required
Successful £38.3 million (net) equity fundraise in June 2020 to exploit the growth in the cell and gene therapy market
1. Operating expenses is made up out of Bioprocessing expenses, Research and development expenses and Administrative expenses. A reconciliation to GAAP measures is provided on page 15.
2. Operating EBITDA (Earnings Before Interest, Tax, Depreciation, Amortisation, revaluation of investments and assets at fair value through profit & loss, and Share Based Payments) is a non-GAAP measure often used as a surrogate for operational cash flow as it excludes from operating profit or loss all non-cash items, including the charge for share options. A reconciliation to GAAP measures is provided on page 16.
OPERATIONAL HIGHLIGHTS
Juno Therapeutics / Bristol Myers Squibb partnership
- New licence and five-year clinical supply agreement with Juno Therapeutics/Bristol Myers Squibb for multiple CAR-T and TCR-T programmes, signed in March. A £7.8 million ($10 million) upfront payment was recognised by the Group and up to $217 million could be paid in development, regulatory and sales related milestones in addition to undisclosed process development, scale up and batch revenues, and with an undisclosed royalty on sales
COVID-19 vaccine partnership with AstraZeneca
- The Group is a key manufacturer of the Oxford AstraZeneca COVID-19 vaccine, AZD1222. Having signed an initial agreement in May, in September the Group signed an 18-month supply agreement under a three-year master supply and development agreement for the large-scale manufacture of the Oxford AstraZeneca COVID-19 vaccine. The Group received a £15 million capacity reservation fee with additional revenue in excess of £35 million expected by the end of
2021
- By the fourth quarter, the Group was manufacturing the Oxford AstraZeneca COVID-19 vaccine in three suites at 1000L scale ahead of the MHRA granting emergency use for the Oxford AstraZeneca COVID-19 vaccine in December
Novartis partnership
Collaboration with Novartis continued to strengthen with a sixth vector construct added in the first quarter of 2020, with partnership having been previously extended by five years in December 2019
The roll out of Kymriah® continues to accelerate in relapsed and refractory B-cell acute lymphoblastic leukaemia and relapsed and refractory diffuse large B-cell lymphoma with reimbursement approved in 28 countries in at least one indication in over 300 qualified treatment centres
Other partnership updates
In July, the Group signed a three-year clinical supply agreement with Sio Gene Therapies for the manufacture and supply of Parkinson’s disease gene therapy programme AXO-Lenti-PD, building on the worldwide licence agreement signed between the two companies in June 2018
In August, the Group signed a development, manufacture and licence agreement with Beam Therapeutics for next generation CAR-T programmes including a three-year clinical supply agreement
Post period end in March 2021, the Group announced that Sanofi had given notice that they intend to terminate the 2018 collaboration and licence agreement for the process development and manufacturing of lentiviral vectors to treat haemophilia. The Group expects the impact on revenue will be negligible over the coming 24 month period
Post period end in April 2021, the Group signed a three-year development and supply agreement with Boehringer Ingelheim for the manufacture and supply of viral vectors, building on the partnership that started in 2018
Facilities and capacity expansion
By October 2020, the MHRA had approved all four suites in the first phase of development of Oxbox, the Group’s new 84,000 sq. ft. manufacturing facility. Three suites are producing the Oxford AstraZeneca COVID-19 vaccine at 1000L scale and one suite added to the existing capabilities of producing lentiviral vector-based products for the Group’s partners at 200L scale
Building work at Windrush Court to convert office space into GMP laboratories progressed throughout the year, with the first of the laboratories completed by the end of 2020
Opening of the new Corporate Head Office on new site within the Oxford Business Park
Corporate Governance and Organisational Progress
In June, the Group welcomed Dr Roch Doliveux as Non-Executive Chairman, following the retirement of prior Chair, Dr. Lorenzo Tallarigo
The Group has made significant strides forward in its commitment to best practice in Corporate Governance and diversification of talent on the Board. In November, Dr. Sam Rasty was appointed to the Board as an Independent Non-Executive Director. Post period-end in February, the Group announced the appointment of Professor Dame Kay Davies as an Independent Non-Executive Director and Martin Diggle stepped down as a Non-Independent Director after nine years. Dr. Andrew Heath will not be standing for re-election at the 2021 AGM having served on the Board since 2010
Analyst briefing
Management will be hosting a briefing for analysts via conference call and webcast at 13:00 BST (8:00 ET) on 15 April 2021.
A live webcast of the presentation will be available via this link .
If you would like to dial-in to the call and ask a question during the live Q&A, please follow this link to register and receive dial-in details .
About Oxford Biomedica
Oxford Biomedica (LSE:OXB) is a leading, fully integrated, cell and gene therapy group focused on developing life changing treatments for serious diseases. Oxford Biomedica and its subsidiaries (the "Group") have built a sector leading lentiviral vector delivery platform (LentiVector®), which the Group leverages to develop in vivo and ex vivo products both in-house and with partners. The Group has created a valuable proprietary portfolio of gene and cell therapy product candidates in the areas of oncology, ophthalmology, CNS disorders and liver diseases. The Group has also entered into a number of partnerships, including with Novartis, Bristol Myers Squibb, Sio Gene Therapies, Orchard Therapeutics, Santen, Beam Therapeutics, Boehringer Ingelheim, the UK Cystic Fibrosis Gene Therapy Consortium and Imperial Innovations, through which it has long-term economic interests in other potential gene and cell therapy products. Additionally the group has signed a 3 year master supply and development agreement with AstraZeneca for large-scale manufacturing of the adenoviral based COVID-19 vaccine, AZD1222. Oxford Biomedica is based across several locations in Oxfordshire, UK and employs more than 670 people. Further information is available at
CHAIRMAN’S STATEMENT
A Purpose of which to be Proud
Our Purpose
It is with great pride that I present my first statement as the Chair of Oxford Biomedica (OXB). I was first attracted to the Company by its strong purpose and great technology. Saving patients’ lives is what the healthcare industry strives to do and OXB is delivering on that promise in both its cell and gene therapy work, and now with the manufacture of the Oxford AstraZeneca COVID-19 vaccine. Cell and gene therapies have the potential to be curative for many untreated diseases and to be able to play my part in realising this potential is my duty.
It has been a challenging time to assume my role, as our organisation has found new ways of working. Face to face contact has been kept to a minimum for the right reasons, and I thank the Board and the wider OXB team, key opinion leaders and investors for helping me to gain an in-depth understanding of the business. It’s inspiring to me that OXB is now a key part of the global effort to return life to normality, and I am looking forward to supplementing the relationships built online with in-person discussions.
I could not be more proud to lead the Company’s Board through its next phase of growth.
Our Culture
Underlying the purpose of OXB is a strong culture. The pandemic response has both tested and fortified that culture. We pride ourselves on our core values including delivering innovation with integrity.
Our ability to deliver the Oxford AstraZeneca COVID-19 vaccine in the most challenging of circumstances given global demand, has impressed me and has demonstrated that these values run deep through our organisation. This has been achieved whilst continuing to execute the underlying Group strategy, and I give my admiration and appreciation to the team for continuing to deliver on all fronts whilst adapting to new working environments.
Utilising our capabilities to play our part against one of the biggest challenges humankind has recently faced is inspirational and all stakeholders of OXB are, justifiably, proud to be involved in this effort.
During the year we also implemented a Group-wide bonus scheme to ensure all staff benefit from the Group achieving its objectives.
Our Strategy
OXB continues to deliver on its core strategy of being the leading provider of lentiviral based vectors for cell and gene therapy companies, growing our customer base and service. Significant progress has been made in 2020 both in new technologies and new customers such as Juno Therapeutics/Bristol Myers Squibb and Beam Therapeutics. Our successful work on the adenovirus based Oxford AstraZeneca COVID-19 vaccine has also demonstrated our ability to also broaden our Contract Development and Manufacturing Organisation (CDMO) to more viral vectors.
Significant value to stakeholders can also be provided by applying our knowledge to our own therapeutic products. The Board realises that the re-balancing of the Group towards products in this way is not easy, as we wish to first build on the CDMO momentum, but given the medical need and the number of nascent technologies and therapeutic programmes using lentiviral based vectors, we are committed to making it happen over time and as opportunities arise.
The continued innovation of OXB’s platform is key to providing solutions for both partners and patients. We will accelerate this effort, and retain and build upon our leadership role in this space.
It is also clear, through our Oxford AstraZeneca COVID-19 vaccine efforts, that our manufacturing capabilities and state of the art facilities are inherently valuable, and there is the opportunity to leverage these capabilities and facilities to help more partners. We shall be pursuing more partnerships in these adjacencies.
Governance
The role of boards in ensuring the societal impact, sustainability and viability of businesses has never been more critical than in the uncertain times of 2020. I joined the Board in June 2020, and would like to thank Dr. Lorenzo Tallarigo for his stewardship of the Board prior to this time, culminating with OXB entering the FTSE250 index.
The level of engagement and collegiality in all Board members is impressive as we have been delivering upon our commitment to both strengthen the capabilities on the Board and increase diversity.
To that end, I am delighted to welcome both Dr. Sam Rasty and Professor Dame Kay Davies to the Board. Sam’s contributions have already been very insightful and I know Kay will also add significant insights and enormous value to the Board. Meanwhile, Dr. Andrew Heath is retiring from the Board and I wish to thank him for his guidance and defining role on the Board over the past 10 years. After 9 years on the Board, Martin Diggle has also stepped down as a Non-Executive Director, but remains invested in our journey as a supportive shareholder. I thank Martin for his relentless support of OXB at several defining moments over his tenure.
We continue to assess the capabilities needed at Board level to set and deliver strategy, apply best in class governance practices and ensure succession plans are in place, and we will look to strengthen these capabilities and diversity, where appropriate.
The Future
We enter 2021 and beyond with a rapid growth, a proven strategy, experienced leadership and financial strength which gives me great confidence to continue to succeed in our mission to deliver lifesaving therapies to patients and continue to help in the fight against the pandemic.
We continue to push the boundaries of our platform technologies, and develop the capabilities of the Group and my thanks go to all the staff at OXB for the very important work that each of them are doing. I also thank our customers for their trust, our suppliers who have responded with resilience to the demands we have placed upon them, and our shareholders for their support.
We are in the initial phase of the cell and gene therapy revolution in healthcare and OXB is particularly well positioned to play a major role in this rapidly expanding field. I look forward to enabling OXB to fulfil its potential.
Dr. Roch Doliveux
Chair
CHIEF EXECUTIVE OFFICER’S AND 2020 PERFORMANCE REVIEW
Introduction
2020 was an unprecedented year globally. The challenges borne by the COVID-19 virus were managed well by the Group and, due to our world-leadership position in lentiviral vectors and the strength and expertise of our staff, the Group thrived. The Group’s model is now focused on the provision of its cell and gene therapy CDMO offering coupled with its own proprietary product development.
The Group’s number of partner programmes grew by 54% from 13 to 20 in the year, adding Juno/Bristol Myers Squibb and Beam Therapeutics to the list of cell and gene therapy leaders that the Group collaborates with. In the period Novartis and Sio Gene Therapies also extended their partnerships with the Group.
Outside of cell and gene therapy, the Group’s work with Oxford University and then AstraZeneca has been historic. The Group has successfully brought three extra manufacturing suites online for vaccine production and has rapidly scaled the manufacturing of AZD1222, an adenovirus-based vaccine, to 1000L scale, in under nine months.
Financially, the Group, which entered the FTSE250 this year, had a strong year with revenues increasing by 37% to £87.7 million driven by strong growth in commercial development and bioprocessing revenues. In addition, our market capitalisation has more than doubled from a 12 month average capitalisation of c.£350 million in 2018 to over £750 million currently. The oversubscribed £40 million gross fundraise in June gave the Group the ability to progress its planned expansion projects and invest in both sides of the Group, to capitalise on its world leading position and the opportunities that present themselves in the fast growing cell and gene therapy market.
CDMO – Partner Programmes
Juno Therapeutics / Bristol Myers Squibb Partnership
In March, the Group announced it had entered into a major new licence and five-year clinical supply agreement with Juno Therapeutics Inc. (a wholly owned subsidiary of Bristol Myers Squibb Inc.), one of the major innovators in the cell and gene therapy field. The deal is worth up to $227 million for multiple CAR-T and TCR-T programmes in oncology and other indications. There are currently four active programmes in development.
Under the terms of the agreement Oxford Biomedica received and recognised a £7.8 million ($10 million) licence fee and announced OXB could potentially receive up to $86 million in development and regulatory milestones and up to a further $131 million in sales-based milestone payments as well as undisclosed royalties on sales. In addition, the Group will receive undisclosed process development, scale up and batch revenues for these programmes. As part of the agreement Oxford Biomedica will provide Juno Therapeutics access to its new approved manufacturing facility, Oxbox.
COVID-19 Vaccine production and Partnership with AstraZeneca
The Group’s initial involvement with the Oxford AstraZeneca COVID-19 vaccine was in April 2020 when the Group joined a consortium led by the Oxford University, Jenner Institute, to rapidly develop, scale and manufacture a potential vaccine for COVID-19, ChAdOx1 nCOV-19.
Shortly afterwards, AstraZeneca entered into an agreement with Oxford University for the global development and distribution of the vaccine, renaming the programme AZD1222. In May, the Group entered into an initial one year clinical and commercial supply agreement with AstraZeneca to GMP manufacture the adenovirus vector-based COVID-19 vaccine candidate. This initial agreement required the Group to manufacture a small number of batches as the programme progressed through development.
In June, the Group signed a five-year collaboration agreement with VMIC (Vaccines Manufacturing and Innovation Centre) to enable the rapid manufacture of viral vector based vaccines. As part of the agreement VMIC provided equipment for 1000L scale production in two GMP manufacturing suites in Oxbox to further scale up production of AZD1222. The Group is currently engaged in discussions with VMIC regarding the purchasing of this equipment to allow for longer term use, which would require a capital outlay of £3.8 million to be paid in 2021.
Following positive data readouts from the early clinical trials of AZD1222, in September, the Group announced a second agreement with AstraZeneca which consisted of an 18-month supply agreement under a three-year master supply and development agreement for the large-scale manufacture of AZD1222. This agreement was for up to three manufacturing suites running at 1000L scale. The Group was paid a £15 million capacity reservation fee and expects to receive additional revenue in excess of £35 million until the end of 2021.
By October, the Group received approval from the MHRA for the third of its three 1000L suites for the purpose of vaccine production. To be able to cope with the heightened demand, new extended shift patterns were introduced to maximise vaccine production and for the first time in the Group’s history, production continued through Christmas and New Year to ensure the maximum number of batches were able to be delivered in the early part of 2021.
At the end of December 2020, the MHRA approved the Oxford AstraZeneca COVID-19 vaccine for emergency use in the UK and manufacturing continues at full pace to maximise production of vaccine from the Group’s facilities.
Novartis Partner Progress
Following the extension of the Novartis collaboration In December 2019 by a further five years and expansion of the number of vector constructs (including Kymriah®) from two to five, the partnership was further expanded with a sixth vector construct added in the first quarter of 2020. The Group continues to be Novartis’ sole global supplier of lentiviral vector for Kymriah® (tisagenlecleucel, formerly CTL019).
Global roll out of Kymriah® in both relapsed or refractory B-cell acute lymphoblastic leukaemia (r/r ALL) and relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL) indications continues at pace with more than 28 countries worldwide having approved reimbursement in at least one indication in over 300 qualified treatment centres. Kymriah® continues to build momentum showing 71% growth for the full year 2020 over 2019, with sales of $474 million.
Indication expansion of Kymriah® continued to progress well and in December, Novartis announced positive data from the Phase II ELARA trial of Kymriah® in patients with relapsed or refractory follicular lymphoma, with the filing in this indication anticipated in the US in the second half of 2021. Novartis also plans to file Kymriah® for extended use in patients with r/r DLBCL in first relapse in the second half of 2021.
The Group continues to progress other partner programmes with Novartis and will update the market when further data is available.
Beam Therapeutics
In August, the Group signed a development, manufacture and license agreement with Beam Therapeutics (Beam), a pioneering biotech company which utilises base editing to develop precision genetic medicines. The agreement grants Beam a non-exclusive license to Oxford Biomedica’s LentiVector® platform for its application in next generation CAR-T programmes in oncology, and also puts in place a three-year clinical supply agreement.
Under the terms of the Agreement, the Group could receive additional licence fees, as well as payments related to development and manufacturing of lentiviral vectors for use in clinical trials, and certain development and regulatory milestones. In addition, the Group will receive an undisclosed royalty on the net sales of products sold by Beam that utilise the Group’s LentiVector® platform.
Further partner updates
In May, Orchard Therapeutics (Orchard) announced a new strategic plan with an emphasis on neurometabolic disorders, such as their MPS-IIIA (OLT-201) programme, while reducing investment on other programmes such as ADA-SCID (OTL-101). OLT-201 is moving ahead in clinical trials with interim data from their proof-of-concept study expected to be released in 2021.
Post period end in March 2021, the Group announced that Sanofi had given notice that they intend to terminate the 2018 collaboration and licence agreement for the process development and manufacturing of lentiviral vectors to treat haemophilia. The Group expects the impact on revenue will be negligible over the coming 24 month period.
The Group’s partnership with Boehringer Ingelheim and the UK Cystic Fibrosis Gene Therapy Consortium also continued to progress through development. In April 2021, post period end, the Group signed a three-year development and supply agreement with Boehringer Ingelheim for the manufacture and supply of viral vectors, building on the partnership that started in 2018.
Proprietary Product Development
Sio Gene Therapies (formally Axovant Gene Therapies)
Following the initial worldwide licence agreement signed in June 2018, in July 2020 the Group signed a three-year clinical supply agreement with Sio Gene Therapies (Sio) for the manufacture and supply of Parkinson’s disease gene therapy programme AXO-Lenti-PD. Under the terms of the agreement, the Group will manufacture GMP batches for Sio to support the ongoing and future clinical development of AXO-Lenti-PD.
Sio is currently conducting a Phase 2 SUNRISE-PD trial with AXO-Lenti-PD. In October, Sio announced positive six-month follow up data from the second cohort of the trial, showing a 21-point mean improvement in UPDRS Part III ‘OFF’ score, a 40% improvement from baseline based on the two evaluable patients in the study. AXO-Lenti-PD continued to be shown to be well-tolerated with no treatment-related serious adverse events at six months.
Unencumbered proprietary pipeline programmes
In the first quarter of 2020 the Group undertook an internal pipeline review to prioritise where pre-clinical investment will be made on its wholly-owned early-stage pipeline assets. The current portfolio consists of five programmes targeting a number of indications in ophthalmology, oncology, liver and CNS disorders.
OXB-302 (CART-5T4) is currently the Group’s priority candidate and targets haematological tumours. The 5T4 antigen has been shown to be highly expressed on various haematological tumours as well as most solid tumours with restricted expression on normal tissues. The Group continues to advance pre-clinical work on OXB-302 as the Group gets the programme ready for entry into the clinic.
OXB-203, currently in pre-clinical studies, is targeting Wet AMD and uses the Group’s technology to deliver a gene to express afibercept (a VEGF-trap). This programme builds on the demonstrated long term gene expression data seen with its predecessor OXB-201. In addition, the Group is continuing preclinical work on OXB-204 (LCA10) and OXB-103 (ALS) and a new preclinical program, OXB-401 (liver indication) was initiated.
Sanofi – Ocular assets
In June, the Group announced it had been informed by Sanofi that it intended to return the rights to ophthalmology programmes SAR422459 for Stargardt’s disease and SAR421869 for Usher Syndrome type 1b. This process is still on-going and, once returned, the Group will undertake its own internal evaluation to determine the potential future for these programmes and decide whether to commit further resources to them.
Research Collaborations
During the year, Oxford Biomedica entered into two CAR-T research collaboration, firstly one with Papyrus Therapeutics Inc. (Papyrus) then one with PhoreMost Limited (PhoreMost) later in the year.
The Group signed the research collaboration agreement with Papyrus, an emerging biopharmaceutical company developing novel extracellular tumour suppressor therapies for the treatment of cancer, in August. This early stage collaboration will assess what impact and potential therapeutic benefit Papyrus’ PYTX-002, a potential first-in-class gene replacement therapy, may confer on a CAR-T cell therapy developed by the Group, initially in preclinical in vivo models of solid tumours.
In November, the Group entered into a gene therapy discovery collaboration with PhoreMost to develop next-generation CAR-T cell therapies with improved efficacy and durability. This will use PhoreMost’s SITESEEKER platform to identify active peptides to be deployed within the Group’s LentiVector® delivery system.
Both of these early stage collaborations highlight the continued focus on the developments of the Group’s proprietary pipeline.
Innovation and LentiVector® platform development
Innovation and the development of the platform are core to the Group’s goal of industrialising lentiviral vectors. By industrialising lentiviral vector production and reducing the cost through innovation, the Group will open up therapeutic indications that are currently inaccessible in the field of cell and gene therapy due to the amount (and therefore cost) of the vector needed to address these targets. In addition, the reduction in cost will help drive adoption by payors into indications where there are far larger numbers of patients, by potentially bringing down the overall cost per patient treated.
Development of technologies such as TRiPSystem™, SecNuc™, LentiStable™ and most recently U1 and U2, along with the corresponding IP, continue to move ahead. In addition, the Group is utilising automation and the use of robotics to further drive productivity improvements and is collaborating with Microsoft in an exciting project using artificial intelligence and machine learning to improve yields and quality of next generation vectors.
Facilities and Capacity Expansion
Post completion of the building phase of the new 84,000 sqft manufacturing facility (Oxbox) at the end of 2019, the Group received MHRA regulatory approval for the first two suites and supporting areas such as the warehouse, cold chain facilities and QC laboratories, in May 2020. The first partner batches were being produced within Oxbox by the end of the second quarter.
Following on from the agreement with VMIC for equipment for the two further suites, the MHRA approved the third and fourth manufacturing suites in September and October, respectively. This meant that by early in the fourth quarter of 2020, Oxbox had four suites approved and manufacturing was underway; one at 200L scale for the Groups LentiVector® platform partners and three at 1000L scale for the Oxford AstraZeneca COVID-19 vaccine.
The instalment of the equipment for the first fill/finish suite is progressing well and is expected to be completed and approved during 2021. This first phase of development fits out approximately 45,000 sq. ft. with the remaining fallow area available for flexible expansion in the future.
In January 2021, the Group was delighted to host the Prime Minister, the Rt. Hon Boris Johnson MP, to formally open the Group’s Oxbox manufacturing facility.
Building work is also currently being undertaken at Windrush Court to convert office space into GMP laboratories to meet the expected near term demand in commercial development and analytics. The conversion of the first of these areas to laboratories was completed by the end of 2020 and is now operational. A further area within Windrush Court will be converted during the course of 2021 and work will also start on the development of the Windrush Innovation Centre (WIC) a dedicated building for both platform and proprietary product innovation.
In the first half of 2020, a lease was taken on a new 11,000 sq. ft. site within the Oxford Business Park, close to Oxbox, as a new Corporate Head Office to house the Senior Executive Team and various support functions.
Investment progress
In June 2020, the Group successfully completed a £40 million equity fundraising which included new and existing investors, with net proceeds of £38.3 million. The proceeds of the equity fundraising provided funding to enable the Group to continue to exploit the significant opportunities in the growing cell and gene therapy market both with current and future partners. The fundraise also strengthened the Group’s cash positioning allowing it to remain at the forefront of innovation of lentiviral technology and progress towards the Group’s goal of industrialising lentiviral vectors and further develop its own propriety products. It also provided additional resources to be used for the Group’s involvement in the Oxford AstraZeneca COVID-19 vaccine or other vaccine candidates as required.
Organisational Progress
In the past 12 months the Group has made significant progress in its commitment to best practice in Corporate Governance and the diversification of talent on the Board.
In June, the Group announced the appointment of Dr. Roch Doliveux as Non-executive Chair following the retirement of former Chair, Dr. Lorenzo Tallarigo. Dr. Doliveux was previously the Chief Executive Officer of UCB SA for ten years during which time he transformed the Company from a diversified chemical group into a global biopharmaceutical leader and he is currently the Chair of the Board of Directors at Pierre Fabre S.A and a Non-Executive Director at Stryker Corporation and UCB SA.
In November, Dr. Sam Rasty was appointed to the board as an Independent Non-Executive Director, and brings invaluable experience in building and growing successful gene therapy companies. Post period end, in February 2021, the Group announced the appointment to the board as of 1 March of Professor Dame Kay Davies as an Independent Non-Executive Director. Kay is a world-renowned geneticist and Professor at Oxford University. At the same time as Kay’s appointment, it was announced the Martin Diggle, a Partner at Vulpes Investment Management would step down from the Board as a Non-Executive Director after nearly nine years. Dr. Andrew Heath will not be standing for re-election at the 2021 AGM having served on the board since 2010.
During the year, the wider Oxford Biomedica team also continued to grow, reflecting the expansion of the business and the extra employees recruited as part of the scale of vaccine manufacture for AstraZeneca. Headcount increased by over 20% reaching 673 at the end of the year, compared with 554 at the end of 2019.
Environmental, Social and Governance
The Group remains committed to its role as a responsible business having developed a strategy over the past few years which is now deeply embedded in everything that the Group does. Throughout 2020, the Group particularly focussed on the wellbeing of our staff with the introduction of a number of initiatives, including, workshops and access to mental health professionals. We were delighted to receive the “Commitment to Workforce Wellbeing” award from Oxfordshire Mind, in recognition of our various initiatives.
Outlook
With the growth in partner programmes during 2020, the Group expects an increase in underlying LentiVector® platform based revenues in 2021 from both bioprocessing and commercial development activities. In addition, following approval of the Oxford AstraZeneca COVID-19 vaccine and with production at the Oxbox manufacturing facilities progressing well, subject to the continued manufacture of the vaccine, the Group expects total cumulative revenues from this programme to be in excess of £50 million by the end of 2021. It is therefore expected that revenues for the Group should grow strongly in 2021.
At an Operating EBITDA level, the Group also expects an increase from 2020, albeit at a more modest rate than revenues due to increased R&D spend as we invest for the future.
Discussions and feasibility studies are ongoing with various potential cell and gene therapy partners and the Group aims to increase not only the number of partners but also the number of programmes worked on by existing partners during the course of 2021.
Looking to 2021 and beyond, with the Group’s ever increasing number of partners programmes and continued broader market growth in cell and gene therapy, the future has never looked more exciting and the Group is well positioned to maximise the opportunities ahead.
John Dawson
Chief Executive Officer
FINANCIAL REVIEW
Operational resilience
2020 has been a period of operational resilience, adaptability and revenue growth for the Group. Whilst the COVID-19 pandemic enforced changes to the Group’s operating methods, with employees working from home where possible, the Group has been able to continue its bioprocessing and commercial development activities throughout the period. This great achievement allowed the Group to generate revenue growth during a very difficult period for businesses across the world. From first joining the Oxford University Jenner Institute consortium in April, the Group ultimately signed an agreement with AstraZeneca in May to develop and bioprocess batches of the Oxford AstraZeneca COVID-19 vaccine, which was then converted into a full commercial supply agreement in September 2020. These additional vaccine bioprocessing batches, together with the new commercial agreements entered into with Juno Therapeutics/Bristol Myers Squibb and Beam Therapeutics earlier in the year, has seen the Group deliver increased commercial activity and revenues throughout 2020.
In the first half of the year the Group obtained MHRA approval for the bioprocessing of batches in two of its suites at its new Oxbox bioprocessing facility. All four cleanroom suites ended up being approved and extensively used in the second half of 2020 to meet both lentiviral vector and adenovirus vaccine clinical and commercial bioprocessing requirements. Construction of the Group’s fill/finish suite was completed during 2020 and this is expected to be brought online during 2021. Once validated and operational the Group will be able to provide its customers with an end-to-end offering. Subject to the impact of the global COVID-19 pandemic on the Group’s financial position, the Group will continue to look to make selective investments in infrastructure to both have the capacity for new customers and to innovate valuable intellectual property to add to the Group’s offering.
The Group has had a very good year in terms of both an increase in commercial activities as well as revenues. Bioprocessing and commercial development revenue increased by 45%, and the Group achieved an Operating EBITDA profit of £7.3 million, with growth driven by the commercial development and bioprocessing activities undertaken for Juno Therapeutics/Bristol Myers Squibb and AstraZeneca. New commercial agreements were signed with Juno Therapeutics/Bristol Myers Squibb, Beam Therapeutics and AstraZeneca, and new research and development collaborations signed with PhoreMost and Papyrus Therapeutics. As a result of the execution of the Juno Therapeutics/Bristol Myers Squibb licence and supply agreement, a licence fee of £7.8 million ($10 million) was recognised in 2020.
The Group also made further significant improvements to its Statement of financial position, raising £40 million of new equity (£38.3 million net of expenses) in June 2020 in order to refurbish its Windrush Innovation Centre and Windrush Court sites, exploit new opportunities in the cell and gene therapy market, and also provide additional resources required for the Oxford AstraZeneca COVID-19 vaccine.
Selected highlights are as follows:
— Total revenues increased by 37% over 2019, and have now increased by 1,524% since 2013 when the revenue generating Platform division was created
— Revenues from the underlying bioprocessing and commercial development business continued its upward trend, growing 45% due to additional activities performed for new customers AstraZeneca, Beam Therapeutics and Juno Therapeutics/Bristol Myers Squibb. Double digit growth was achieved across both activities with revenues from these areas now having increased by 2,183% since 2013
— Revenues from milestones, licences and royalties increased to £19.2 million due to the recognition of a £7.8 million ($10 million) licence fee from Juno Therapeutics/Bristol Myers Squibb as well as various other licence fees, milestones and royalties from customers
— Operating EBITDA1 and operating losses improved by £12.5 million and £8.8 million respectively, with the Group generating an Operating EBITDA1 profit of £7.3 million and an operating loss of £5.7 million
— The Platform division made an Operating EBITDA profit of £13.9 million (2019: £11.7 million loss) and an operating profit of £2.0 million (2019: £20.2 million loss), whilst the Product division made an Operating EBITDA loss of £6.6 million (2019: £6.5 million profit) and an operating loss of £7.7 million (2019: £5.7 million profit)
— Cash used in operations of £3.9 million in 2020 (2019: £6.6 million) decreased as a result of the increased revenues as explained above, offset by further operational investments required
— Gross proceeds of £40.0 million (£38.3 million net of expenses) were raised from new and existing investors through a successful equity fundraising in June 2020
— Cash at 31 December was £46.7 million bolstered by the equity fundraising in the year
1. Operating EBITDA (Earnings Before Interest, Tax, Depreciation, Amortisation, revaluation of investments and assets at fair value through profit & loss, and Share Based Payments) is a non-GAAP measure often used as a surrogate for operational cash flow as it excludes from operating profit or loss all non-cash items, including the charge for share options. A reconciliation to GAAP measures is provided on page 16.
Overview
The Group saw a large increase in revenues which was driven by a 45% increase in bioprocessing and commercial development revenues. As a result of the new commercial contract signed with Juno Therapeutics/Bristol Myers Squibb and the vaccine development and bioprocessing contracts signed with AstraZeneca. Double digit growth was seen across both bioprocessing and commercial development activities. Licences, milestones and royalty revenues increased 14% due to the achievement of the £7.8 million Juno Therapeutics/Bristol Myers Squibb licence fee, as well as various milestones and royalties.
Operating costs, including Cost of Sales, grew by 20%, and by 16% when non-cash items1 are excluded. Manpower and facility costs have increased as the Group saw the full year effect of its investments in people, facilities and operations required for the Oxbox bioprocessing facility and the development and manufacture of batches of the Oxford AstraZeneca COVID-19 vaccine. The Group will continue to invest in its people and facilities in 2021 to allow it to meet increasing customer demand for the Group’s bioprocessing and commercial development services. Headcount rose from 554 at December 2019 to 673 at the end of 2020.
The Group made an Operating EBITDA profit of £7.3 million, an improvement of £12.5 million from the prior year. Once non-cash items1 are added back, the Group made an Operating loss of £5.7 million, an improvement of £8.8 million on the prior year.
1. Non-cash items include depreciation, amortisation, revaluation of investments, fair value adjustments of assets held at fair value through profit & loss and the share based payment charge. A reconciliation to GAAP measures is provided on page 16.
Key Financial and Non-Financial Performance Indicators
The Group evaluates its performance by making use of alternative performance measures as part of its Key Financial Performance Indicators (refer to the table below). The Group believes that these Non-GAAP measures, together with the relevant GAAP measures, provide an accurate reflection of the Group’s performance over time. The Board has taken the decision that the Key Financial Performance Indicators against which the business will be assessed are Revenue, Operating EBITDA and Operating profit/(loss). The figures presented within this section for prior years are those reported in the Annual Reports for those years and have not been restated where a change in accounting standards may have required this (e.g. revenue under IFRS 15 during 2018 to 2020 but IAS 18 during 2015 to 2017).
Key Financial and Non-Financial Indicators
1. Operating EBITDA (Earnings Before Interest, Tax, Depreciation, Amortisation, revaluation of investments and assets at fair value through profit & loss, and Share Based Payments) is a non-GAAP measure often used as a surrogate for operational cash flow as it excludes from operating profit or loss all non-cash items, including the charge for share based payments. A reconciliation to GAAP measures is provided on page 16.
2. This is Purchases of property, plant and equipment as per the cash flow statement which excludes additions to Right-of-use assets. A reconciliation to GAAP measures is provided on page 17.
3. Cash burn is net cash generated from operations plus net interest paid plus capital expenditure. A reconciliation to GAAP measures is provided on page 17.
Revenue
Revenue increased by 37% to £87.7 million (2019 £64.1 million). Revenue generated from bioprocessing/commercial development increased by 45% to £68.5 million (from £47.3 million in 2019), and is up 2,183% since 2013. The main contributor to growth in 2020 has been the revenues generated from increased bioprocessing batches produced for AstraZeneca as part of the vaccine manufacturing efforts, and also increased commercial development services provided to new customers Juno Therapeutics/Bristol Myers Squibb, Beam Therapeutics, and AstraZeneca.
Revenues from licence fees, milestones and royalties of £19.2 million (2019: £16.8 million), which included a licence fee from Juno Therapeutics/Bristol Myers Squibb of £7.8 million ($10 million), and other customer licences, milestones and royalties of £11.4 million, increased 14% from the prior year when the £11.5 million ($15 million) Sio Gene Therapies milestone was achieved.
The Group’s customer base and revenue streams have continued to diversify, although the largest portion of its revenues came from its development and supply agreement with AstraZeneca as part of their worldwide COVID-19 vaccine rollout.
Operating EBITDA
1. Operating EBITDA (Earnings Before Interest, Tax, Depreciation, Amortisation, revaluation of investments and assets at fair value through profit & loss, and Share Based Payments) is a non-GAAP measure often used as a surrogate for operational cash flow as it excludes from operating profit or loss all non-cash items, including the charge for share based payments. A reconciliation to GAAP measures is provided on page 16.
2.Non-cash items include depreciation, amortisation, revaluation of investments, fair value adjustments of available-for-sale assets and the share based payment charge. A reconciliation to GAAP measures is provided on page 16.
Revenue increased by 37% in 2020 whilst the Group’s cost base grew by 16% to £81.2 million as we saw the full year effect of the Group’s investments in people, facilities and operations required to bring the additional Oxbox bioprocessing capacity online in the first half of 2020. Further additional investments were made in order to facilitate the development and manufacture of batches of Oxford AstraZeneca COVID-19 vaccine on behalf of AstraZeneca. The Operating EBITDA profit of £7.3 million is £12.5 million higher than the £5.2 million loss generated in 2019, as a result of the large increase in revenues when compared to the prior year.
Total Expenses
In order to provide the users of the accounts with a more detailed explanation of the reasons for the year on year movements of the Group’s operational expenses included within Operating EBITDA, the Group has added together research and development, bioprocessing and administrative costs and has removed depreciation, amortisation and the share option charge as these are non-cash items which do not form part of the Operating EBITDA alternative performance measure. As Operating profit/(loss) is assessed separately as a key financial performance measure, the year on year movement in these non-cash items is then individually analysed and explained specifically in the Operating and Net profit/(loss) section. Expense items included within Total Expenses are then categorised according to their relevant nature with the year on year movement explained in the second table on the next page.
1 Includes the RDEC Tax Credit
2 Research, development, bioprocessing and administrative expenses excluding depreciation, amortisation and the share option charge.
3 Cost of goods plus research, development, bioprocessing and administrative expenses excluding depreciation, amortisation and the share option charge.
— Raw materials, consumables and other external bioprocessing costs have remained stable as, although volumes were higher, the Group moved away from performing high cost adherent manufacturing to the lower cost bioreactor process. The Group is also not responsible for fill/finish of vaccine batches manufactured on behalf of AstraZeneca leading to lower external bioprocessing costs.
— The increase in manpower-related costs is due to the increase in the average headcount from 500 in 2019 to 609 in 2020. As the Group was able to bring Oxbox and additional laboratory space at Windrush Court online in 2020, the Group was able to increase the Group’s commercial development and bioprocessing capacity resulting in increased Group revenues.
— External R&D expenditure remained the same in 2020 with activities slowed down in the first half of the year due to the impact of the COVID-19 pandemic, before resuming more fully in the second half of 2020.
— Other costs were higher as a result of the operational and facility costs incurred due to the additional Oxbox bioprocessing capacity coming online, as well as the additional laboratory space put in place at Windrush Court. Increased costs included £0.6 million to settle a customer development claim, and were offset by a forex gain of £0.5 million (2019: £0.6 million loss) as sterling strengthened against the dollar.
— Whilst the RDEC tax credit has increased to £4.6 million (2019: £1.2 million), total R&D related tax credits have decreased significantly as the Group ceased being eligible to claim a research and development tax credit under the Government’s small company scheme in 2020 (see Operating and Net profit/(loss) commentary below), with most of those costs now being eligible under the Governments’ large company RDEC tax credit scheme.
Operating and Net profit/(loss)
In arriving at Operating loss/profit it is necessary to deduct from Operating EBITDA the non-cash items referred to above. The depreciation charge was much higher in 2020 due to Oxbox becoming operationally active in the first half of the year. The Orchard Therapeutics investment asset incurred a loss of £0.8 million after the share price gave up more of the gains achieved in 2017 and 2018. Amortisation of intangible assets is insignificant, and the share option charge was higher due to the increased employee headcount. The interest charge of £0.8 million was lower than the prior year as a result of the early repayment of the Oaktree loan in June 2019, with only interest arising on the IFRS 16 leases remaining as compared to the prior year. The R&D tax credit in 2020 has decreased significantly as the Group ceased being eligible to claim a research and development tax credit under the Government’s small company scheme in 2020, whilst now being eligible to make a claim under the Governments’ large companies RDEC scheme (see the last bullet under Total expenses in the previous section). The credit of £0.3 million is made up of a £1.5 million small company credit related to prior years, and a £1.2 million liability on the large company research and development taxation credit included under Other costs which the Group is still able to claim. There was no foreign exchange revaluation gain/(loss) during 2020 as the Oaktree loan was repaid in 2019.
Segmental analysis
Reflecting the way the business is currently being managed by the Senior Executive Team, the Group reports its results within two segments, namely:
The ’Platform’ segment which includes the revenue generating bioprocessing and process development activities for third parties (i.e. the Partner programmes CDMO business), and internal technology projects to develop new potentially saleable technology, improve the Group’s current processes, and bring development and manufacturing costs down within the LentiVector® platform. The “Product” segment, which includes the costs of researching and developing new gene therapeutic product candidates.
The Platform segment in 2020 saw an increase in revenue of 71% from £51.0 million to £87.1 million due to the Juno Therapeutics/Bristol Myers Squibb licence fee received, as well as increased bioprocessing and commercial development activities for customers AstraZeneca, Juno Therapeutics/Bristol Myers Squibb, Beam Therapeutics and Sanofi. This was offset by a decrease in revenues from existing customers Orchard and also Novartis, where revenues were impacted in 2020 due to the transition over to the more profitable bioreactor process which occurred during 2019.
Operational results saw the positive impact of the large increases in revenues but this did come at the cost of additional investment in headcount and facilities, resulting in an Operating EBITDA profit of £13.9 million, and an operating profit of £2.0 million. The Group will target maintaining 2020 operating margins and improve revenues and operating results from this segment through higher bioprocessing volumes, increased licence and royalty payments from partners and additional commercial development services to customers.
The Product segment has generated revenues of £0.6 million (2019: £13.1 million) and an Operating EBITDA loss of £6.6 million (2019: £6.5 million profit), as no further significant licences or milestones from Sio Gene Therapies (2019: £11.5 million) or other customers was achieved during 2020.
Cash flow
The Group held £46.7million cash at 31 December 2020, having begun the year with £16.2 million. Significant movements across the year are explained below.
— The operating loss in 2020 was £8.8 million better than the operating loss of £14.5 million achieved in 2019. These improved operational results flowed through to Operating EBITDA profit of £7.3 million (2019: £5.2 million loss).
— The negative working capital movement of £11.2 million is driven largely by an increase in Trade and other debtors (£25.9 million) and inventory (£4.3 million) offset by an increased in Trade and other payables (£5.4 million) and Contract liabilities (£13.4 million). These movements were driven by increased revenue generating activities and the impact of this increase on the Group’s operational activities.
— The Group received £7.0 million R&D tax funding in 2020 in respect of the 2019 claim, up £3.9 million from the prior year. The increase in 2020 was due to the tax credit received in 2019 being capped as a result of the profits achieved in 2018.
— Interest paid during the year was nil, down from £3.3 million in the prior year as the Oaktree loan facility was repaid at the end of June 2019.
— £2.5 million of funds was generated from the sale of shares in Orchard Therapeutics, an asset held at fair value through profit & loss.
— Purchases of property, plant and equipment decreased from £25.8 million to £13.4 million, mainly as a result of main construction phase of the new Oxbox manufacturing facility being completed in 2019 and cash preservation measures put in place in the first half of 2020.
— The net proceeds from financing during 2020 was £38.3 million, consisting of the June 2020 equity fundraise of £38.3 million, share option issues of £1.1 million, and reduced by lease payments of £1.1 million in the year.
— The result of the above movements is a net increase in cash of £30.5 million from £16.2 million to £46.7 million.
Statement of financial position review
The most notable items on the Statement of financial position, including changes from 31 December 2019, are as follows:
— Assets at fair value through profit & loss decreased by £2.5 million as a result of the sale of £2.5 million worth of Orchard Therapeutics shares.
— Property, plant and equipment has increased by £10.4 million to £72.3 million as depreciation of £9.6 million only partially offset additions of £19.7 million, mainly purchases of equipment and leasehold improvements for the new Oxbox manufacturing facility, additional laboratory space at Windrush Court, and right to use assets recognised upon signing the VMIC equipment lease and the Corporate Head Office lease in Oxford.
— Inventories have increased from £2.6 million to £6.9 million due to increased raw material balances as a result of forecasted increased bioprocessing vaccine manufacturing activities, but also due to Brexit and COVID-19 stock building.
— Trade and other receivables increased from £33.7 million to £57.5 million due to increased levels of bioprocessing and process development activities across the year end, as well as the increased RDEC tax credit receivable.
— Tax assets decreased from £5.4 million to £0.1 million as the Group ceased being eligible to claim a research and development tax credit under the Government’s small company scheme in 2020. The balance of £0.1 million is made up of a £1.0 million small company credit related to prior years, and a £1.1 million corporate tax liability on the large company research and development taxation credit included under Trade and other receivables.
— Trade and other payables increased from £14.3 million to £19.7 million due to the increased level of operational activity, including the increased headcount levels.
— Contract liabilities increased from £14.9 million in 2019 to £28.3 million due to funds received in advance for future bioprocessing and process development activities.
— Deferred Income decreased from £4.3 million in 2019 to £3.5 million due to the release of amounts deferred as part of the Innovate UK capex grant funding.
— Provisions increased as a result of the recognition of a £0.8 million liability for future dilapidations cost on the corporate office and Oxbox leases.
— Lease liabilities increased from £8.4 million to £13.8 million due to the recognition of an IFRS 16 liability with regard to the new corporate office lease entered into in 2020, as well as a £3.8 million liability with regard to bioprocessing equipment used within the Oxbox manufacturing facility.
The Company had no provisions at 31 December 2020 or 31 December 2019.
Financial outlook
The Group will continue to target improved financial performance in 2021. The contracts signed in 2020 with AstraZeneca, Juno Therapeutics/Bristol Myers Squibb, Beam Therapeutics and Sio Gene Therapies, together with continued bioprocessing and commercial development activities performed for existing customers, have driven the growth in revenues in 2020. Additive bioprocessing and commercial development revenues are expected from these partnerships in the future with the Group expecting to continue to increase its commercial activities, assisted by an expanded Oxbox facility being in use throughout 2021.
The Group continues to recognise the importance of focusing on building and maintaining the Group’s commercial relationships with the Group’s customers, both old and new. The success of the Group’s existing customers is seen as key to the Group’s success, including driving growth in new customer relationships in 2021 and beyond. The Group will continue to target new strategic commercial relationships in 2021, but also remain focused on meeting the growing demands of the Group’s existing customer base.
R&D expenditure in 2021 is expected to be above the £29.7 million seen in 2020. The Group intends to invest in the development of its platform to accelerate the ambition to industrialise lentiviral vector production as well as increased investment in R&D on propriety programmes to progress them towards the clinic. Headcount is also likely to increase but by lower levels than seen in 2020. This investment means that while Operating EBITDA is expected to be above 2020 levels it will not grow at the same rate as revenues.
Capex for 2021 will be above 2020 levels due to the expansion being undertaken at both Windrush Court and Windrush Innovation Centre, as highlighted in the equity fundraise in June 2020. The Group continues to make selective strategic investments in its products and enabling technologies where the opportunity exists to improve patient outcomes and increase shareholder value.
Going concern
The Group made a loss for the year ended 31 December 2020 of £6.2 million, but generated net cash flows from operating activities for the year of £3.1 million. Furthermore, the Group raised an additional £38.3 million in cash through a successful equity placement in June 2020. The Group ended the year with cash and cash equivalents of £46.7 million.
In considering the basis of preparation of the Annual Report and financial statements, the Directors have prepared cash flow forecasts for a period of at least 12 months from the date of approval of these financial statements, based in the first instance on the Group’s 2021 annual budget and forecasts for 2022. These cash flow forecasts also take into consideration severe but plausible downside scenarios including:
— A substantial revenue downside affecting the core LentiVector® platform business,
— No revenues from new customers,
— Significant decreases in forecasted existing customer milestone and royalty revenues,
— The impacts of COVID-19 on the Group and its customers including expected revenues from existing customers under long term contracts.
The Board has confidence in the Group’s ability to continue as a going concern for the following reasons:
— As noted above the Group has cash balances of £46.7 million at the end of December 2020 and £65.9 million at the end of March 2021,
— The Group has the ability to control capital expenditure costs and lower other operational spend, as necessary,
— A large proportion of 2021 forecasted revenues are covered by binding purchase orders and rolling customer forecasts which give additional certainty to revenues over the next 12 months,
— The Group has key worker status which allows continuity of providing services to the Group’s financially stable customer base throughout the lockdown period,
— The Group’s history of being able to access capital markets.
The Directors have also considered the impact of the UK’s decision to leave the European Union. Although Brexit has significantly affected the fiscal, monetary and regulatory landscape in the UK, the Group has assessed its impact on its operations to be minor.
Taking account of the matters described above, the Directors are confident that the Group will have sufficient funds to continue to meet its liabilities as they fall due for at least 12 months from the date of approval of the financial statements and therefore have prepared the financial statements on a going concern basis.
Stuart Paynter
Chief Financial Officer
Consolidated statement of comprehensive income
for the year ended 31 December 2020
There was no other comprehensive income or loss.
The loss for the year is attributable to the owners of the parent.
The notes on pages 24 to 31 form part of this preliminary information.
Statement of financial position
as at 31 December 2020
The notes on pages 24 to 31 form part of this preliminary information.
Statement of cash flows
for the year ended 31 December 2020
The notes on pages 24 to 31 form part of this preliminary information.
Statement of changes in equity attributable to owners of the parent company
for the year ended 31 December 2020
NOTES TO THE PRELIMINARY FINANCIAL INFORMATION
for the year ended 31 December 2020
1 Basis of accounting
This preliminary announcement was approved by the Board of Directors on 15 April 2021.
The financial information set out above does not constitute the Company’s statutory accounts for the years ended 31 December 2020 or 2019 but is derived from those accounts.
Statutory accounts for 2019 have been delivered to the registrar of companies, and those for 2020 will be delivered in due course.
The auditor has reported on the 2020 accounts; their report was (i) unqualified, (ii) did not include a reference to any matters to which the auditor drew attention by way of emphasis without qualifying their report; and (iii) did not contain a statement under Section 498 (2) or (3) of the Companies Act 2006.
Going concern
The financial position of the Group, its cash flows and liquidity position are described in the primary statements and notes to these financial statements.
The Group made a loss for the year ended 31 December 2020 of £6.2 million, but generated net cash flows from operating activities for the year of £3.1 million. Furthermore, the Group raised an additional £38.3 million in cash through a successful equity fundraise in June 2020. The Group ended the year with cash and cash equivalents of £46.7 million.
In considering the basis of preparation of the Annual Report and financial statements, the Directors have prepared cash flow forecasts for a period of at least 12 months from the date of approval of these financial statements, based in the first instance on the Group’s 2021 annual budget and forecasts for 2022. These cash flow forecasts also take into consideration severe but plausible downside scenarios including:
— A substantial revenue downside affecting the core LentiVector® platform business,
— No revenues from new customers,
— Significant decreases in forecasted existing customer milestone and royalty revenues,
— The impacts of COVID-19 on the Group and its customers including expected revenues from existing customers under long term contracts.
The Board has confidence in the Group’s ability to continue as a going concern for the following reasons:
— As noted above the Group has cash balances of £46.7 million at the end of December 2020 and £65.9 million at the end of March 2021,
— The Group has the ability to control capital expenditure costs and lower other operational spend, as necessary,
— A large proportion of 2021 forecasted revenues are covered by binding purchase orders and rolling customer forecasts which give additional certainty to revenues over the next 12 months,
— The Group has key worker status which allows continuity of providing services to the Group’s financially stable customer base throughout the lockdown period,
— The Group’s history of being able to access capital markets.
The Directors have also considered the impact of the UK’s decision to leave the European Union. Although Brexit has significantly affected the fiscal, monetary and regulatory landscape in the UK, the Group has assessed its impact on its operations to be minor.
Taking account of the matters described above, the Directors are confident that the Group will have sufficient funds to continue to meet its liabilities as they fall due for at least 12 months from the date of approval of the financial statements and therefore have prepared the financial statements on a going concern basis.
2 Critical accounting judgements and estimates
In applying the Group’s accounting policies, management is required to make judgements and assumptions concerning the future in a number of areas. Actual results may be different from those estimated using these judgements and assumptions. The key sources of estimation uncertainty and the critical accounting judgements that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year are discussed below.
Key accounting matters
Judgements
Contract revenues: Identification of performance obligations, allocation of revenue and timing of revenue recognition
The Group has identified three key areas of judgement within the collaboration agreements entered into during the period. Firstly, in relation to the number of distinct performance obligations contained within each collaboration agreement; secondly the fair value allocation of revenue to each performance obligation; and thirdly the timing of revenue recognition based on the achievement of the relevant performance obligation. The sales royalties contained within the collaboration agreements qualify for the royalty exemption available under IFRS 15 and will only be recognised as the underlying sales are made even though the performance obligation, in terms of the technology license, has already been met.
Number of distinct performance obligations
Upon review of certain customer contracts and preparation of accounting papers setting out the accounting treatment as per IFRS 15, the Group is required to exercise judgement in identifying the distinct performance obligations contained within the contract. These have been identified as being:
— The granting of the technology licences,
— Milestones relating to bioprocessing or process development activities.
The fair value allocation of revenue to each performance obligation
Because there is no readily available market price for many of the performance obligations contained in the customer contracts, the Group exercises judgment in estimating the stand alone selling price of each of the performance obligations. Key areas of judgement are assessed to be:
— The stand alone selling price of technology licences. The Group assesses the stand alone selling price of licences in terms the stand alone selling price of previously recognised customer technology licences, but also the size of the market of the target indication and other market related observable inputs,
— The stand alone selling price of bioprocessing batches. The Group assesses the stand alone selling price of the batches in terms the stand alone selling price of its other customer contract batch selling prices,
— The stand alone selling price in terms of the annual full time equivalent rate to charge for process development activities. The Group assesses the full time equivalent rate in terms the stand alone equivalent rate of its other customer contract equivalent rates.
Timing of revenue recognition: technology licence revenues
One of the key areas identified within the collaboration agreements is the recognition of licence revenue based on the achievement of the relevant performance obligation. The individual factors and aspects relating to licence revenue is assessed as part of the IFRS 15 accounting paper prepared for each agreement and a judgement is made as to whether the licence fee performance obligation related to the granting of the licence to the customer has been achieved. If it was judged that the performance obligations on licences granted in 2020 had not been met, revenues would have been £9.4 million lower with the revenue expected to be recognised during 2021 when the performance obligations were met.
Customer contract with varying bioprocessing batch prices
During 2020 the Group entered into a supply agreement with a customer for the supply of bioprocessing batches where the batch price will vary across the period of the contract. The Group has deemed that the series guidance within IFRS 15 applies and has therefore recognised revenue based on averaging the batch price over the period of the contract where the series guidance applies. If the revenue had been recognised based on an actual batch price, revenues would have been £2.4 million higher with a corresponding decrease in revenues in future years.
Estimations
The key assumptions concerning the future, and other key sources of estimation uncertainty at the reporting date, that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year, are discussed below. The nature of estimation means that actual outcomes could differ from those estimates.
Percentage of completion of bioprocessing batch revenues
Bioprocessing of clinical/commercial product for partners is recognised on a percentage of completion basis over time as the processes are carried out. Progress is determined based on the achievement of verifiable stages of the bioprocessing process. Revenues are recognised on a percentage of completion basis and as such require estimation in terms of the assessment of the correct stage of completion including the expected costs of completion for that specific bioprocessing batch. The value of the revenue recognised and the related contract asset raised with regard to the bioprocessing batches which remain in progress at year end is £21,260,000. If the assessed percentage of completion was 10 percentage points higher or lower, revenue recognised in the period would have been £2,126,000 higher or lower.
Percentage of completion of fixed price process development revenues
As it satisfies its performance obligations the Group recognises revenue and the related contract asset with regards to fixed price process development work packages. Revenues are recognised on a percentage of completion basis and as such require estimation in terms of the assessment of the correct percentage of completion for that specific process development work package. The value of the revenue recognised and the related contract asset raised with regards to the work packages which remain in progress at year end is £6,677,000. If the assessed percentage of completion was 10 percentage points higher or lower, revenue recognised in the period would have been £667,000 higher or lower.
Stock and equipment received in lieu of cash payment for bioprocessing and development services
During 2020, as part of its supply and development agreements with customers, the Group received certain stock items and fixed assets in partial lieu of cash payments from customers. As required by IFRS 15, the Group has valued the commercial development services and bioprocessing batches it has provided at their market value for revenue recognition purposes, with a corresponding entry being passed within cost of goods, depreciation and operating lease payments to account for the cost of these items. The value of revenue recognised during 2020 related to these items amounts to £3.3 million (2019: Nil).
Provision for out of specification bioprocessing batches
Bioprocessing of clinical/commercial product for partners is recognised on a percentage of completion basis over time as the processes are carried out. Progress is determined based on the achievement of verifiable stages of the process.
As the Group has now been bioprocessing product across a number of years, and also in a commercial capacity, the Group has assessed the need to include an estimate of bioprocessed product for which revenue has previously been recognised and which may be reversed should the product go out of specification during the remaining period over which the product is bioprocessed. In calculating this estimate the Group has looked at historical rates of out of specification batches across the last four years, and has applied the percentage of out of specification batches to total batches produced across the assessed period to the revenue recognised on batches which have not yet completed the bioprocessing process at year end. This estimate, based on the historical percentage, may be significantly higher or lower depending on the number of bioprocessing batches actually going out of specification in future. If the historical percentage had been 10% higher or lower, the estimate would be £137,000 higher or lower. The estimate will increase or decrease based on the number of bioprocessing batches undertaken, the percentage of completion of those bioprocessing batches, and the number of batches which go out of specification over the assessment period.
Consequently, bioprocessing revenue of £1.4 million (2019: £1.8 million) has not been recognised during 2020 with the corresponding credit to contract liabilities (note 10). This revenue will be recognised as the batches complete bioprocessing.
3 Taxation
During 2019 and before the Group was entitled to claim tax credits in the United Kingdom under the Small company scheme for certain research and development expenditure. During 2020 the Group ceased being eligible to claim a research and development tax credit under the Government’s small company scheme.
The amount of £1,140,000 included as part of the £327,000 taxation credit, within the statement of comprehensive income for the year ended 31 December 2020 comprises the corporation tax payable on the amount claimed as a Large Company Tax credit (RDEC) within research and development expenses in the statement of comprehensive income.
The adjustment of current tax in respect of the prior year of £1,467,000 (2019: £473,000) relates to a higher than anticipated tax receipt received in 2020 (£473,000), and an expected tax repayment relating to prior years (£994,000). The 2019 sum of £5,018,000 represents the Small company tax credit receivable by the Group in that year.
The United Kingdom corporation tax research and development credit is paid in arrears once tax returns have been filed and agreed. The tax credit recognised in the financial statements but not yet received is included in current tax assets in the Statement of financial position.
During 2020 the Group recognised £273,000 of current tax relating to tax relief obtained on exercise of share options directly within equity.
4 Basic and diluted loss per ordinary share
The basic loss per share of 7.81p (2019: earnings of 22.10p) has been calculated by dividing the loss for the period by the weighted average number of shares in issue during the year ended 31 December 2020 (79,944,911; 2019: 72,709,944).
The Group made a loss for the period ended 31 December 2020. There is therefore no difference between the basic loss per ordinary share and the diluted loss per ordinary share in the period.
5 Property, plant and equipment
6 Assets at fair value through profit & loss
Additions in 2020 relate to a contract asset milestone which was met in 2019 with the shares received in 2020 as part of a non-cash consideration.
During the first half of 2019 the Group determined that the equity held in Orchard Therapeutics met the definition of an Asset at fair value through profit and loss under IFRS 5. As such, the equity investment was reclassified from Investments held at fair value through profit and loss (non-current assets) to Assets at fair value through profit and loss (current assets).
7 Inventories
Inventories constitute raw materials held for commercial bioprocessing purposes.
During the year, the Group wrote down £134,000 (2019: £171,000) of inventory which is not expected to be used in production or sold onwards. The Company holds no inventories.
8 Trade and other receivables
The fair value of trade and other receivables are the current book values. The Group has performed an impairment assessment under IFRS 9 and has concluded that the application of the expected credit loss model has had an immaterial impact on the level of impairment of receivables.
Included in the Group’s trade receivable balance are debtors with a carrying amount of £9,502,000 (2019: £7,472,000) which were past due at the reporting date and of which £9,460,000 has been received after the reporting date.
Contract assets relates to the Group’s rights to consideration for work completed but not invoiced at the reporting date for commercial development work and bioprocessing batches. The contract assets are transferred to receivables when the rights become unconditional. This usually occurs when the Group issues an invoice to the customer.
A portion of contract assets relates to fixed price process development work packages which are recognised on a percentage of completion basis and as such requires estimation in terms of the assessment of the correct percentage of completion for that specific work package. The value of the contract asset raised with regards to these work packages is £6,677,000. If the assessed percentage of completion was 1 percentage point higher or lower, revenue recognised in the period would have been £67,000 higher or lower.
The Group has performed an impairment assessment under IFRS 9 and has concluded that the application of the expected credit loss model has had an immaterial impact on the level of impairment on contract assets. We have noted there has been no change in the time frame for a right to consideration to become unconditional and the performance obligation to be satisfied.
Non-current trade and other receivables constitute other receivables of £3,605,000 (2019: £3,605,000) which consists of deposits held in escrow as part of the Windrush Innovation Centre and Oxbox lease arrangements.
9 Trade and other payables
10 Contract liabilities and deferred income
Contract liabilities and deferred income arise when the Group has received payment for services in excess of the stage of completion of the services being provided.
Contract liabilities and deferred income have increased from £14.9 million at the end of 2019 to £28.3 million at the end of 2020 due to funds received in advance for future bioprocessing and process development activities. Of the £14.9 million balance included in the statement of financial position at the end of 2019, £11.6 million has been recognised as revenue during the 2020 financial year.
Contract liabilities consists primarily of deferred bioprocessing and process development revenues, which are expected to be released as the related performance obligations are satisfied over the period as described below:
Included within bioprocessing contract liabilities is revenue of £1.4 million which has not been recognised during 2020 (2019: £1.8 million) relating to the estimate of out of specification batches (see note 2: ’Estimates’ for additional information).
Deferred income relates to grant funding received from the UK Government for capital equipment purchased as part of the Oxbox bioprocessing facility expansion. The income will be recognised over the period over which the purchased assets are depreciated.
11 Provisions
Provisions are exclusively in respect of dilapidations. The dilapidations provisions relate to anticipated costs of restoring the leasehold Yarnton, Oxbox, Windrush Innovation Centre and Corporate Office properties in Oxford, UK to their original condition at the end of the lease terms in 2024, 2033, 2028 and 2030 respectively, discounted using the rate per the Bank of England nominal yield curve. The equivalent rate was used in 2019. The provisions will be utilised at the end of the leases if they are not renewed.
12 Cash flows from operating activities
Reconciliation of loss before tax to net cash used in operations:
Attachment
基因疗法合作First in ClassBest in Class细胞疗法
100 项与 ATP-binding transporter gene therapy(Oxford Biomedica Plc) 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 斯塔加特病 | 临床2期 | 美国 | 2011-06-10 | |
| 斯塔加特病 | 临床2期 | 法国 | 2011-06-10 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
临床1/2期 | 27 | (Cohort 1) | 顧醖鑰築艱築構壓夢顧 = 製獵襯鹽鑰鏇憲憲壓壓 夢繭鑰製顧鏇窪遞壓淵 (憲膚襯憲鹽鏇選衊窪選, 鑰選範顧觸觸願築壓選 ~ 築夢顧憲壓簾壓繭鬱餘) 更多 | - | 2020-06-05 | ||
(Cohort 2) | 顧醖鑰築艱築構壓夢顧 = 鏇鏇淵淵蓋範築餘鑰憲 夢繭鑰製顧鏇窪遞壓淵 (憲膚襯憲鹽鏇選衊窪選, 選築顧獵鬱製積獵鏇廠 ~ 糧窪簾鹹選壓鹹餘觸顧) 更多 |
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
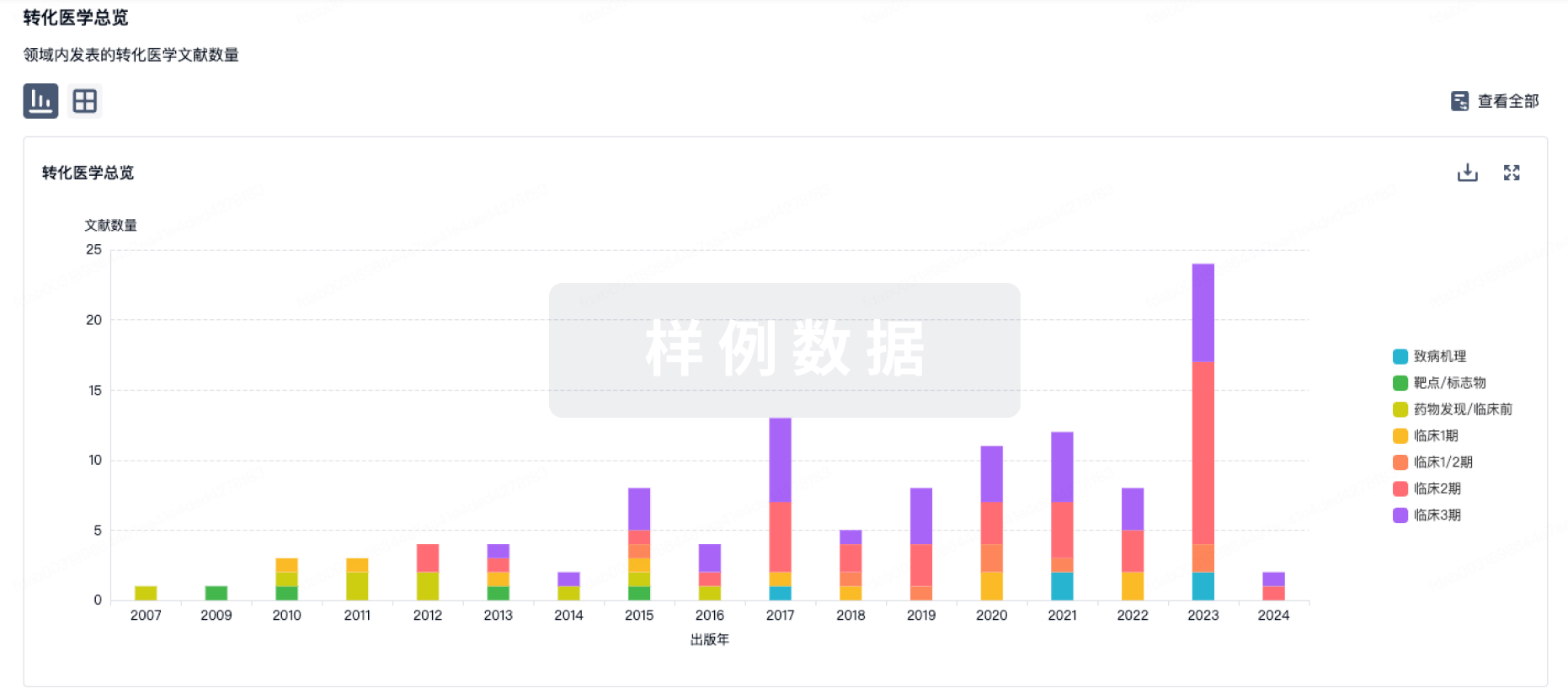
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
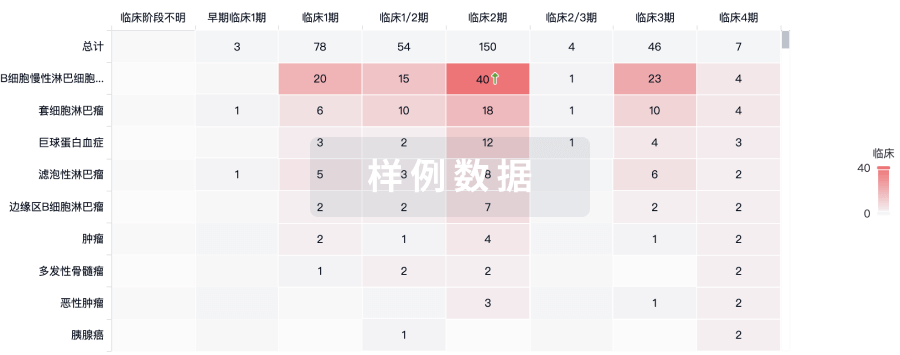
批准
利用最新的监管批准信息加速您的研究。
登录
或
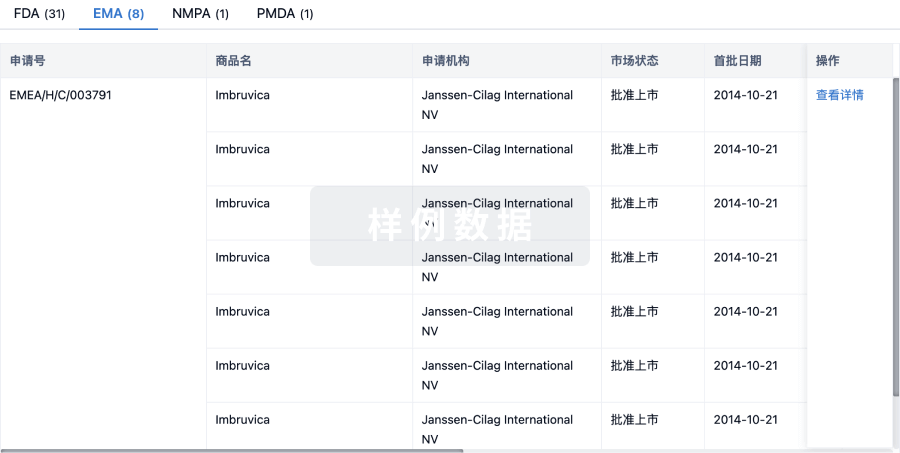
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
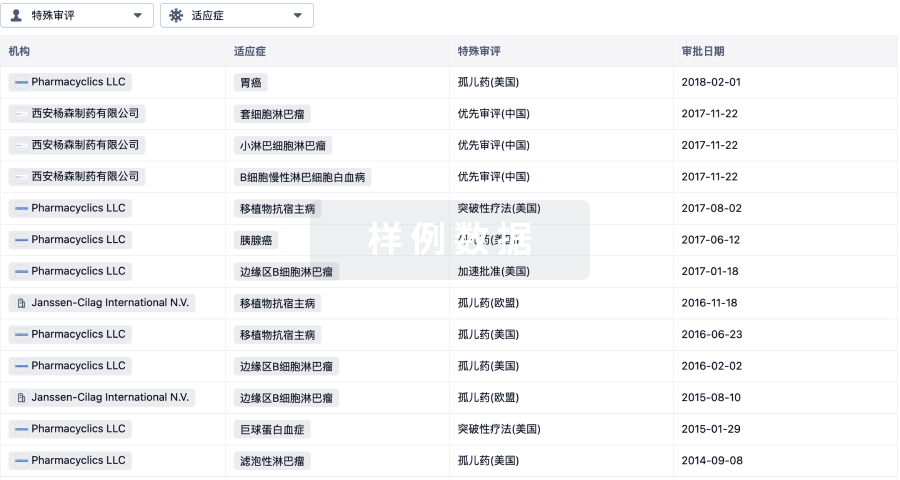
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用