更新于:2024-05-01
International Society for Pharmaceutical Engineering, Inc.
更新于:2024-05-01
概览
关联
100 项与 International Society for Pharmaceutical Engineering, Inc. 相关的临床结果
登录后查看更多信息
0 项与 International Society for Pharmaceutical Engineering, Inc. 相关的专利(医药)
登录后查看更多信息
3
项与 International Society for Pharmaceutical Engineering, Inc. 相关的新闻(医药)2024-03-26
▲ 金斯瑞蓬勃镇江新厂房开幕论坛将于2024年4月18-19日盛大启幕,点击图片了解更多活动资讯生物药行业是一个充满活力和快速发展的领域,主要涉及生物医药产品的研发、生产和销售。行业的发展受到全球健康需求、科技进步和政策支持的推动,同时作为高风险、强监管行业,持续稳定地生产出满足患者需要、符合法规和注册要求的药物一直是制药行业的基础与命脉,而国际化药品行业的监管更是一个复杂的过程,涉及到多个层面,包括国家法规、国际组织、药品安全、有效性和质量控制等方面。随着国内生物制药近几年的快速发展,很多企业的临床产品即将进入注册申报阶段以及商业化生产阶段。我们将结合生物制药行业的特点、近年来国际化的监管趋势及金斯瑞蓬勃生物多年的实践经验,探讨生物制药企业如何适应国际化市场监管的要求。 01计算机化系统促进和提升质量管理体系药品研发和生产企业作为直接影响患者生命健康安全和国计民生的行业,质量管理在企业的实际工作中发挥着非常重要的作用。将传统的质量管理体系和现代化的计算机系统相融合,充分发挥计算机系统的优势,促进和提升传统的质量管理体系将会成为医药行业发展的必然趋势。计算机化系统通过先进的科学技术手段和准确的算法可以更加高效、准确的确保质量管理体系的健康运行,并且能够提前预警和及时警示质量管理体系运行中的一些异常,更有利于提升企业数据可靠性管理水平和防止人为差错等数据可靠性管理中出现的问题使企业的综合质量管理水平更上一个台阶。为了让质量管理体系更加合规、高效、透明,更好的服务于客户以及满足监管的期望和要求,金斯瑞蓬勃生物从人员培训、文件管理、物料管理、实验室管理到质量管理等引入了涵盖各主要业务部门的计算机化系统,如:TMS, DMS, SAP, LIMS, QMS等。在引入计算机系统的同时,我们也建立和总结了一套完善的计算机化系统管理的体系和经验,如:完整的计算机化系统生命周期的管理(参见图一)、从传统的CSV(计算机化系统验证)升级科学的CSA(计算机系统保证)(参见图二)以及设置专门的GMP IT团队等。图一 计算机化系统生命周期图二 从传统的CSV升级为CSA 02深入掌握国际法规和标准随着CMC研究的不断推进,对药物的安全性、有效性、疗效等了解加深,GMP管理要求也越来越加强。基于对国内外药品监管法规和标准的深入研究和理解,如NMPA、FDA、EMA、ICH、PDA、ISPE等国家、地区、行业的要求,建立适用于不同阶段的质量管理体系(PAC Guideline),既能保证合规,又能避免法规符合性的不足与浪费。PAC QMS设计原理及优势如图三所示: 图三 PAC QMS设计原理和优势从发现和工艺开发经不同的临床阶段到工艺验证批和商业化生产,是一个相当长的过程。随着产品阶段的推进,工艺的不断成熟,对人员/患者影响风险的增大,对药物安全性和有效性的要求不断加强,其GMP的监管和质量管理的强度也应存在差异化策略。下面以检测仪器的管理为例介绍下不同阶段质量管理体系强度上的差别,以起到抛砖引玉的作用:在早期开发阶段,检测数据的准确性主要是指导研究成果,对人员的健康安全几乎没有影响,所以在此阶段检测仪器管理的重点是校准。随着项目的推进,到了临床阶段,由于药物需要进入人员/患者体内以及实验人群数量的增加,对人员的健康安全影响逐步增大,所以在此阶段检测仪器管理除了校准,额外需要关注检测仪器的确认,如安装确认和运行确认等。到了工艺验证(PPQ)及商业化阶段,随着工艺的锁定、检测方法的完善以及工艺验证和后续批量生产的要求,在此阶段检测仪器的管理除了校准、确认外需要有完善的全生命周期的管理流程,如:完整的设备确认包括性能确认以及维护保养计划等完全的管理要求。金斯瑞蓬勃生物自2019年起,建立了国际化的适用于不同阶段的质量管理体系并在实际运用中持续完善提升。在该体系运营保证下,我们已成功完成了中美欧等共80个以上的IND批件。 03加强员工培训与技能提升培养一支具备国际化视野和专业技能的团队是构建国际化生物药质量体系的关键。加强员工培训,提高员工的质量意识和操作技能,鼓励员工参与国际交流和合作,拓宽视野,提升技能。每年基于实际执行情况,开展针对性的差距分析、专题研讨、专项知识培训,辅以周期性的年度知识强化培训,同时邀请行业专家针对行业热点话题进行针对性培训、法规解读、现场检查与指导等。 04加强项目质量要求的管理和保证建立固定且程序化、标准化的客户项目质量要求管理程序,将客户的每一个需求,无论是书面协议还是会议决定等通过书面流程及时、准确和清晰的传递到每一个责任部门并严格落实,是CDMO企业的首要责任,也是药品全生命周期中(包括研发、临床和商业化阶段)面临的挑战。我们通过多年的实践经验为每一个客户项目自承接起就建立一套独立、完整的CQV (Client Quality Verification) 管理平台,确保客户的每一个期望和要求准确无误的传递给相关部门和执行人员,设置专人定期检查确认且项目交付时再逐一复核确认,执行完整的闭环管理。 05持续改进与优化通过定期收集和分析各项数据、客户反馈、各类审计(包括自检)等信息,识别(潜在)问题,并进行适当的调查、风险评估及各级管理会议回顾,分析问题并确定适当措施,持续优化质量管理体系。同时,关注行业动态和技术发展趋势,及时引入新技术和工艺,提升生产效率和产品质量。 06注重质量文化的培育在公司内部营造注重质量、追求卓越的文化氛围。通过“多培训、多宣传、多鼓励”等,提高全体员工对质量重要性的认识,形成企业共同的“质量价值观—质量是每个人的质量”以实现公司的“质量愿景—通过最佳的实践以及最差状况的规划来保持一流的全球质量体系,为患者提供质量最好的产品”。 07关于金斯瑞蓬勃生物通过多年的经验、良好的实践和持续改进,我们建立了具有金斯瑞蓬勃生物特色的涵盖药品研发至商业化的不同阶段的全生命周期的药品质量管理体系,在合理的成本控制下高标准的满足不同国家和地区的制药法规和客户的要求。更好的满足客户需求,构建和日益完善满足国际监管要求的质量体系是我们一直努力的方向。目前金斯瑞蓬勃生物已顺利获得《药品生产许可证》;多次零缺陷通过欧盟QP远程或现场审计;已为来自国内外的客户获得超过80个全球IND批件;100%通过全球十几个国家/地区的近200家客户审计和多家外部专业机构的商业化GMP审计,包括来自美国的知名专业机构。这些数据足以证明蓬勃生物的质量体系已达到国际先进的质量体系标准,同时向客户和合作伙伴展示了公司的实力和信誉。点击视频,观看最新厂房动画:点击"阅读原文",进入官网报名~
2023-06-14
引用本文贾晓艳,李素梅,陈跃武,刘继峰,何国强*.国内外防止交叉污染的药品共线生产质量管理法规和监管模式的比较[J].中国食品药品监管.2023.03(230):72-77.国内外防止交叉污染的药品共线生产质量管理法规和监管模式的比较Comparison of National and International Quality Management Regulations and Regulatory Modes for Collinear Production of Drugs to Prevent Cross Contamination贾晓艳香港奥星集团JIA Xiao-yanAUSTAR Group李素梅黑龙江省药品审核查验中心LI Su-meiCentre for Drug Inspection of Heilongjiang陈跃武香港奥星集团CHEN Yue-wuAUSTAR Group刘继峰香港奥星集团LIU Ji-fengAUSTAR Group何国强*香港奥星集团MARS Ho*AUSTAR Group摘 要Abstract交叉污染是指不同原料、辅料及产品之间发生的相互污染。当使用共用设施设备生产不同药品时,潜在的交叉污染就成为了一项极其关键的风险管控问题。近年来,全球许多药品监管机构或组织针对共用设施、共线生产质量管理以及产生的交叉污染,更新发布了相关法规和指南。但目前,共用设施下的交叉污染及相关缺陷项目(如多产品共线评估、清洁验证有效性等)依然高频出现在其发布的GMP 检查不符合项中。本文从药品共线生产质量管理基本原则,即法律法规要求、药品上市许可持有人主责、产品的生命周期和清洁验证的生命周期,以及质量风险管理等方面,论述了全球不同药品监管机构或组织对药品共线生产质量管理和交叉污染的监管要求,并汇总形成了相应的管理流程和方法,以供相关监管人员和药品生产企业人员参考。Cross contamination is the mutual contamination of different raw materials, excipients and finished products.Potential cross contamination is a very critical risk control issue when different drug products are manufactured in shared facilities. In recent years, different regulatory authorities or organizations have issued and updated the regulations and guidelines against shared facilities, collinear production management and cross contamination. Cross contamination in shared facilities and related defect items (such as collinear production assessment and cleaning validation) still frequently appear in the Good Manufacturing Practice (GMP) non-conformance items issued by many drug regulatory agencies or organizations around the world. This paper discussed the regulatory requirements of different regulatory authorities or organizations on cross contamination and collinear production management mainly from the following aspects: requirements of laws and regulations, drug marketing authorization holder’s principle responsibility, product lifecycle, cleaning validation lifecycle and quality risk management. Besides, it also summarized relevant management procedures and methods, expecting to provide reference for relevant regulators and personnel of drug-manufacturing enterprises.关键词Key words共线生产管理;交叉污染;风险评估collinear production management; cross contamination; risk assessment鉴于已知的药品产品特性与风险, 世界卫生组织(World Health Organization,WHO)、药品检查合作计划(Pharmaceutical Inspection Co-operation Scheme,PIC/S)以及中国、美国、欧盟等国家和地区的药品监管机构要求使用专用或隔离的独立厂房设施设备生产部分特殊产品,包括高致敏性药品(如青霉素类)、生物制品(如卡介苗或其他用活性微生物制备而成的药品)、β- 内酰胺结构类药品,以及某些激素类、细胞毒性类、高活性化学药品[1-3]。此外,如何在药品共线生产质量管理活动中有效识别多产品共线生产的交叉污染风险(如生产设施、系统、设备等共用风险),共线生产质量管理体系需要包含的关键项目,以及必须采取的合适的风险控制和缓解措施等,始终是其GMP 关注的基本方面。我国国家药品监督管理局食品药品审核查验中心于2023 年3 月6 日发布了《药品共线生产质量风险管理指南》[4](以下简称《指南》)。《指南》的发布并非“新”的或是“额外”的要求,而是依托我国现行GMP 要求,结合我国现阶段药品共线生产质量管理实际制定的一份细化文件。《指南》明确了药品共线生产质量管理需要遵守的法律法规及规范要求,明确了药品上市许可持有人(marketing authorization holder,MAH)主责原则、生命周期原则、质量风险管理原则、风险控制措施与收益整体平衡原则,并要求构建一套完整的基于产品知识、工艺知识的药品共线生产质量管理体系。广大药品生产企业应当结合国内外药品监管机构或组织对共线生产质量管理的要求,建立完善的、贯穿药品全生命周期的共线生产质量管理体系,采取有效的防止交叉污染的控制措施,更好地保障药品生产安全。一、国内外药品共线生产质量管理需要遵守的法律法规要求笔者梳理了中国、美国、欧盟药品监管机构以及PIC/S 对于交叉污染和共线生产质量管理的监管要求如下。我国GMP 第四十六条明确了对药品共线生产的要求,强调了多产品共线生产评估要点,包括药品的特性、工艺、预定用途、厂房设施设备等[1]。美国现行GMP 为《动态药品生产管理规范》(Current Good Manufacture Practices,CGMP), 其中第C 子部分《厂房和设施》(Buildings and Facilities) 第211.42 节《设计与建造特点》(Design and Construction Features)(c)部分明确了对药品生产操作区域、隔离区域、相关控制系统等的要求,以防止在操作过程中发生污染或混淆[3]。欧盟GMP 第一部分《药品生产的基本要求》(Basic Requi rements for Medicinal Products)第三章《厂房与设备》(Premises and Equipment)3.6项规定,所有产品均应当通过恰当的设计与制造设施操作来避免交叉污染;预防交叉污染的措施应当与风险相适应;应当使用质量风险管理基本原则来评估与控制风险[4]。PIC/S 发布的043-1《共享设施交叉污染检查备忘录》(Aide-Memoire Cross-Contamination In Shared Facilities)提出,应根据所处理材料的危害性确定风险控制措施,并通过由GMP、质量控制(quality control,QC)、质量风险管理(quality riskmanagement,QRM) 三者有机结合的质量保证系统(quality assurance system,QAS)正确实施;充分记录控制措施并监测其有效性,并在考虑科技进步的情况下进行定期审查;交叉污染风险管理体系必须与患者保护明确关联起来[5]。笔者结合上述针对交叉污染和共线生产质量管理的监管要求,总结形成了多产品共线生产可行性评估决策树,如图1 所示。除了决策树中描述的情形,即便经过风险评估得出可以共线生产的结论,也仍会有不可接受的风险,还需要进一步评估共线生产的可行性。例如,对于疫苗类产品,原液制备要求设施设备独立,灌装工序可以共线生产,但是用于共线生产的设备的关键部件、器具等是否专用则仍需要进一步评估论证。二、国内外药品共线生产质量管理需要依据的原则(一)药品上市许可持有人主责原则MAH 和受托药品生产企业应当按照法律法规要求,建立共线生产质量管理体系。双方的主要职责分工应在委托合同和质量协议中明确说明,如图2 所示。国家药监局发布的《药品上市许可持有人检查要点(征求意见稿)》针对共线生产评估提出“持有人应当对受托生产企业共线生产风险控制措施的有效性进行定期审核,若共线生产品种发生变更,需重新进行风险评估”的要求[6]。美国食品药品监督管理局(FDA)行业指南《药品委托生产安排:质量协议》(Guidance for Industry: Contract Manufacturing Arrangements for Drugs: Quality Agreements)规定,如果受托方为多个所有者生产药品,质量协议应指出各方要如何沟通关于防止交叉污染和维护可追溯性[7]。欧盟GMP 第一部分《药品生产的基本要求》(Basic Requi rements for Medicinal Products) 第七章《外包活动》(Outsourced Activities)规定,委托方有最终责任确保通过相关程序使外包活动受控[3]。因此,针对受托方的共线生产活动,MAH 的管理职责应包含评估、审核、批准以及定期审核、回顾、变更与风险评估等,且相关内容应在委托合同和质量协议中体现。受托方应根据委托合同和质量协议中的职责内容积极执行。(二)生命周期原则药品共线生产质量管理策略中提及的生命周期涵盖药品研发、技术转移、药品生产、上市后监测四个阶段。针对上述各阶段,共线生产质量管理需要重点进行的活动与数据收集工作如图3 所示。值得关注的是,共线生产质量管理中的清洁验证是针对药品生命周期开展的,即将清洁验证分为清洁工艺设计和开发、清洁工艺验证、持续清洁工艺确认三个阶段。1. 清洁工艺设计和开发在共线生产的场景下,清洁工艺设计和开发阶段是清洁验证工作的重点。其典型活动包括收集积累药品药理学和毒理学数据,如基于健康的暴露限度(healthbased exposure limit,HBEL)数据,制订清洁工艺开发方案,选择合适的清洗剂,建立清洁剂残留分析方法、清洁操作规程以及相应的可接受标准。2. 清洁工艺验证清洁工艺验证主要包括确认设备、公用工程与系统状态,创建清洁工艺验证方案(包括选择取样点、取样方法等),开展相关工作人员培训,执行清洁工艺验证方案并形成最终的验证报告。3. 持续清洁工艺确认持续清洁工艺确认主要包括建立定期评审制度,确定清洁工艺取样和检测的扩展范围,监控清洁工艺能力,审核偏差和变更项目。在清洁验证方面, 国际制药工程协会(International Society for Pharmaceutical Engineering,ISPE) 发布的指南《清洁验证生命周期》(Cleaning Validation Lifecycle)[8] 可供药品生产企业参考。在药品生命周期的各个阶段,药品生产企业均应确保质量管理体系有效运行并实现持续改进, 尤其是针对生命周期中的变更管理。在共线生产的场景下,药品生产企业应建立标准作业程序(standard operating procedure,SOP),对评估、批准和实施建议的变更(如新产品引入、产品规格发生变化等)进行描述和管理;对变更管理活动有清晰的职责描述(涉及MAH、受托药品生产企业等);对实施变更的后续措施进行检查(如在每批产品生产结束后进行残留检测)与回顾,最终确认变更对于共线生产质量管理是有效的。(三)质量风险管理原则ICH Q9《质量风险管理》(Quality Risk Management)[9] 概述了一个持续迭代的风险管理过程,包括危害的识别、分析和评估,以及对相关风险的控制策略。该指南鼓励在药品生命周期内采用基于科学和风险的方法。在开展共线生产风险评估时,基于质量风险管理的原则,使用风险管理工具评估潜在的风险,并通过风险评估将所识别和分析的风险与给定的风险标准进行对比,确定影响拟共线生产品种安全性和质量的潜在因素。ISPE 基准指南《基于风险的药品生产》(Risk-Based Manufacture of Pharmaceutical Products)[10] 提供了一系列基于风险分析的方法,以管理所有级别药品生产过程中交叉污染的风险, 从而保证将交叉污染维持或低于可接受限度。该指南初始关注点是GxP(Good “x” Practice,良好管理规范)提到的交叉污染内容,且对工业卫生(涉及操作人员安全)相关内容也进行了充分说明。药品生产企业在计划实施共线生产质量管理时, 应当基于质量源于设计(Quality by Design,QbD) 的原则, 在厂房设施设计阶段通过对拟共线生产品种的GMP 监管因素可行性和环境健康安全(environment health safety,EHS)领域的职业健康卫生因素可行性进行分析,对潜在风险进行识别分析并采取充分的控制措施,以及充分建立防止交叉污染的控制措施, 保持交叉污染的风险在可接受水平以下。欧盟GMP 第一部分《药品生产的基本要求》(Basic Requi rements for Medicinal Products) 第五章《生产》(Production) 的5.21 部分明确表示:“应根据质量风险管理过程的结果来决定用于控制交叉污染风险所需的技术和组织方面的措施。”[3] 因此,药品生产企业应从技术层面和组织层面对共用设施交叉污染控制措施进行持续改进和完善。在技术层面,密闭技术是药品生产过程中防止污染和交叉污染的重要措施,密闭设备在控制交叉污染方面起着重要作用,有望成为制药行业的应用热点。根据共线生产品种的危害程度,ISPE 发布的良好实践指南《强效化合物密闭》(Containment for Potent Compounds)[11] 推荐了以下控制措施。一级密闭是指从实际生产设备中减少物质的扩散的措施,主要包括:为压片机增加外壳和附加设备(如除尘单元);用于活性药物成分(active pharmaceutical ingredient,API)称重的隔离器;局部排风系统。二级密闭指的是减少在主要密闭隔离范围以外的物质扩散的措施,主要包括:洁净室;在生产区和走廊之间的气锁系统和压差。在组织层面,清洁工艺开发和验证仍然是有效控制共线生产设施交叉污染的重要研究方向。对于计划开展药品共线生产的企业而言,共线生产风险控制相关内容都需要被纳入到质量管理体系相关文件中,如清场、变更管理、纠正和预防措施(corrective action and preventive action,CAPA)、培训和日常监测等。计划开展药品共线生产的企业应当在项目早期(如概念设计阶段),就开始考虑共线生产质量管理的总体质量策略,并设计建立设备密闭验收标准,在项目交付运营之前建立共线生产质量管理体系,从而控制与厂房设施设计、设备系统选型、日常监测等方面相关联的交叉污染风险。(四)风险控制措施与收益整体平衡原则药品生产企业人员普遍认为,风险几乎是不能被完全消除的,风险管理的参与者只能尽量减少风险。部分企业人员已经认识到,在进一步采取措施控制风险的过程中,当风险降低到一定程度时可能会达到收益递减点。因此,他们会保留那些对所分析的生产活动影响不大的低水平风险,以求生产出符合质量要求的产品。这些风险被归类为“低至合理可行”(as low as reasonably practicable,ALARP)[12], 企业人员会根据风险及收益分析情况来选择是否接受。一般情况下,针对高风险无需考虑成本,应尽可能降低风险;而对于接近可接受区域的风险,在技术与经济平衡方面则有较大的灵活性。需要注意的是,药品生产企业人员在没有采取进一步的控制措施或没有进行正式的风险及收益分析时,不应当接受不可容忍的风险。三、对药品共线生产质量管理的思考和建议基于上述对国内外共线生产质量管理监管要求的分析和比较,建议药品生产企业制订共线生产质量管理总体规划,并建立适宜的风险评估方法和控制措施,以确保交叉污染风险得到有效控制。具体措施和建议包括:为药品共线生产策略的设计、实施及改进提供分析和指导;基于质量风险管理的理念,理解药品共线生产可能存在的风险,以及暴露和风险的关系;科学确定残留可接受限度;建立残留物的分析方法;分析产生交叉污染的途径;采取降低交叉污染的措施;持续监控交叉污染水平。四、结语目前,国内外许多药品监管机构和组织都对药品共线生产的交叉污染风险给予了高度关注,要求在GMP 检查过程中关注药品生产企业是否采取了针对交叉污染的风险管理措施。药品生产企业应当制订共线生产质量管理策略并加以文件化,以确定药品是否可以使用共用设施设备生产,同时应根据风险评估的输出结果确定必要的控制措施,对共线生产的交叉污染风险进行管控,科学、合理地提供有效的共线生产管理措施。同时,建议药品监管部门重点关注药品生产企业在共线生产方面采取的实际控制措施和具体执行情况。第一作者简介贾晓艳,硕士,香港奥星集团。专业方向:质量风险管理、验证管理通讯作者简介何国强,香港奥星集团,行政总裁。专业方向:制药工艺和技术参考文献请扫描二维码《中国食品药品监管》杂志国际标准连续出版物号:ISSN 1673-5390国内统一连续出版物号: CN 11-5362/D期刊级别: 国家级 刊期:月刊《中国食品药品监管》作为国家药品监督管理局机关刊,创刊于1998年,目前是国家药品监督管理局主管的隶属于中国健康传媒集团的科学性、专业性核心期刊;更是研究和宣传中国食品药品监管政策、建立科学监管理念、提高监管水平、服务我国食药产业健康发展的重要平台。点击封面订阅杂志喜欢就请点个"在看"吧~
2023-05-05
·药研
由香港奥星集团与药研学院共同推出的第100期直播公开课《信息化技术的合规与PAT技术在口服固体制剂中应用》将于5月18日19:00开播,约2小时。PAT技术是基于对制药工艺的深刻理解,借助先进的检测工具,通过数据建模与分析确定关键工艺参数、优化设计空间,并进行工艺终点判断。PAT技术覆盖产品研发、中试到商业化生产全过程,通过PAT获取数据实施过程监测,确保产品质量持续稳定,有助于识别产品质量风险,制定和实施高效的控制策略。本次信息化技术的合规与PAT技术在口服固体制剂中应用课题,将分享信息化技术的合规要求与建设路径,PAT技术与实践案例两个方面的主要内容,以期满足智能制造背景下日益严格的监管要求,为企业的产品质量稳定可控、高效生产赋能。一、直播摘要课题一:信息化技术的合规应用与实施路径分析1. 信息化技术的合规新要求2. 信息化/智能化发展趋势与发展机遇3. 实施信息化前企业需要进行的准备工作,需要具备的条件4. 信息化技术实施路径与建设要求5. 案例分析6. 答疑课题二:PAT技术在口服固体制剂中的应用1. 连续制造与PAT技术2. PAT技术在OSD中的应用3. NIR技术及其应用4. 在线粒径分析技术及其应用5. 案例分析6. 答疑二、主讲人柯争先奥星集团丨确认和验证咨询总监、高级工程师18年+制药行业项目管理、调试&确认及设施管理维护经验,对数字化合规、计算机化系统验证、确认与验证、数据完整性、GEP有深入研究。所参与项目曾获ISPE年度设施奖;参与药监局、ISPE、行业协会、大学、媒体机构等培训及论坛交流活动百余次;为制药企业客户提供培训累计数百场次。参与编写或整理《制药行业质量风险管理:实践指南》、《密闭合规资料集》、 《细胞治疗产品合规资料集》、 《制药工艺验证实施手册》、《欧盟GMP/GDP法规汇编(中英文对照版)》、《制药行业制造执行系统实施手册》、《全球数据可靠性法规指南汇编(中英文对照版)》、《ASTM E2500应用指南》等多部书籍和资料集。在《流程工业》、《机电信息》、《中国医药报》、《弗戈工业媒体》等刊物及媒体发表了数十篇的制药合规领域专业论文。蔡兴诗奥星集团丨口服固体制剂工艺专家近10年处方工艺研发及工艺放大技术转移经验,熟悉口服固体制剂各工艺流程,DOE实验设计经验,以及丰富工艺建模与PAT建模经验。专注制药行业QbD理论实践,以及药品研发、生产工艺测试、PAT技术、热熔挤出工艺、湿法制粒线、多功能流化床包衣、干法制粒等应用技术的研究。三、免费观看方式1.手机扫码观看:2.电脑观看:将下方链接复制到电脑浏览器即可观看https://qqy.h5.xeknow.com/sl/2lHYrx四、有奖转发转发本直播到朋友圈满3小时/或集赞8个/或分享到3个100人以上微信群,截图加助理审核并提供邮箱,即可在直播结束后获得《CDE、CFDI等专家文章合集550+篇》。务必备注单位+姓名【关于香港奥星集团】香港奥星集团是一家科技型制药工程解决方案提供商,业务范围遍及全球50 多个国家和地区,与世界领先的制药公司合作,致力于保护和提升人类健康。集团拥有全面的制药行业技术知识,在洁净公用工程、制药自动化和信息化、制药配方工艺科技、生物工艺和技术、合规&卓越运营、实验室科技和设施、生物安全科技和设施、清洁、杀菌、消毒、洁净室/HVAC/EMS/BMS、质量/测量及分析、灌装、冻干及检测、密闭科技领域有领先的技术应用能力和高品质设备和系统。香港奥星集团成立于1991年,总部设于中国北京,2014年11月7日,成功在香港联合交易所挂牌上市(股票代码:6118)。集团拥有多个在国内外市场占据领先地位的业务和产品品牌,包括H+E Pharma 、BIOSYSTEC、 STERIS-AUSTAR、ROTA-AUSTAR、Pharma LeanTec、Pharma LeanDigital、AUSMILL、Aunity、 Purvita等。香港奥星集团官网:https://www.austar.com.cn/直播合作:手机/微信 15911172616
细胞疗法
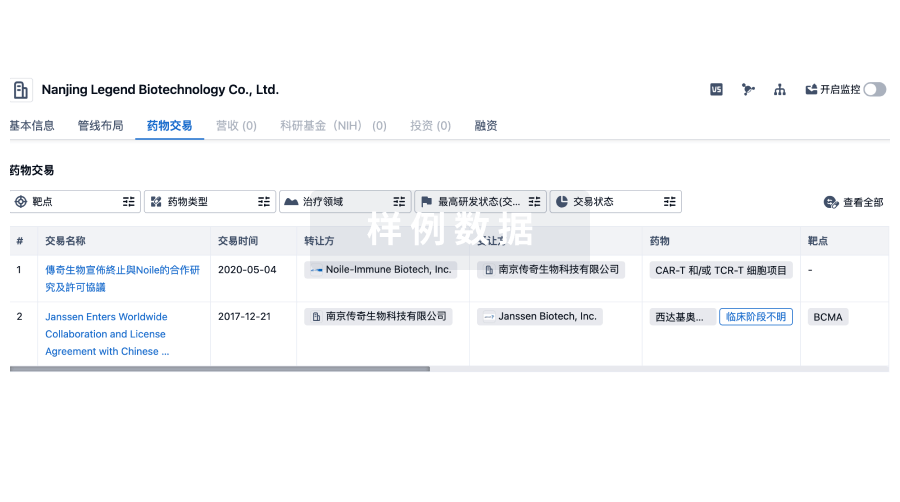
100 项与 International Society for Pharmaceutical Engineering, Inc. 相关的药物交易
登录后查看更多信息
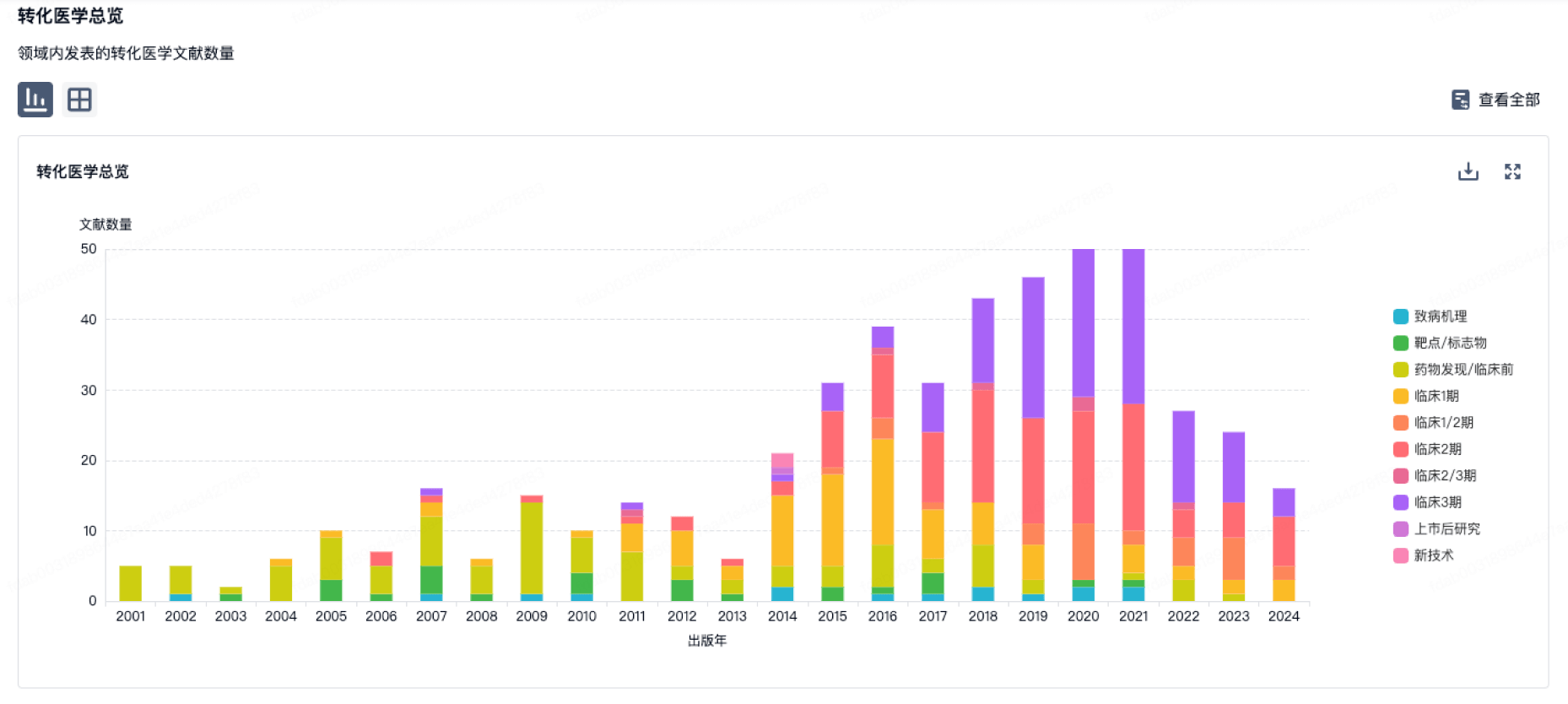
100 项与 International Society for Pharmaceutical Engineering, Inc. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年07月03日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
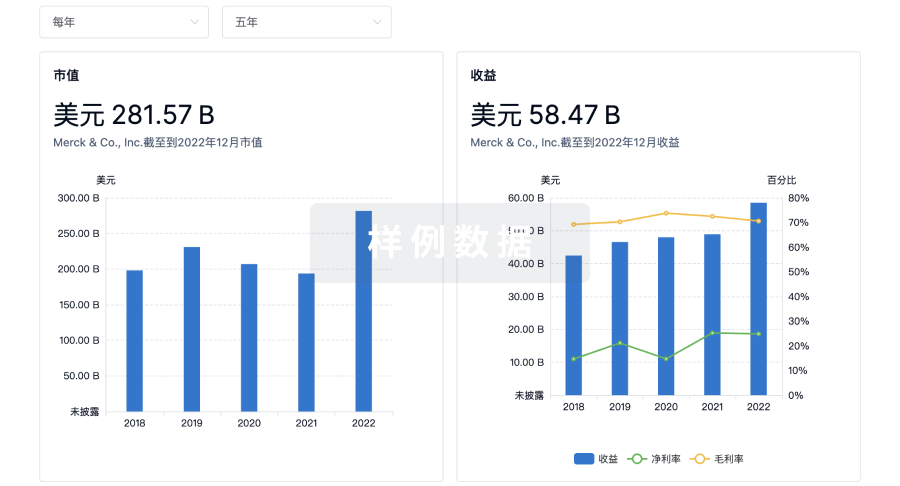
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
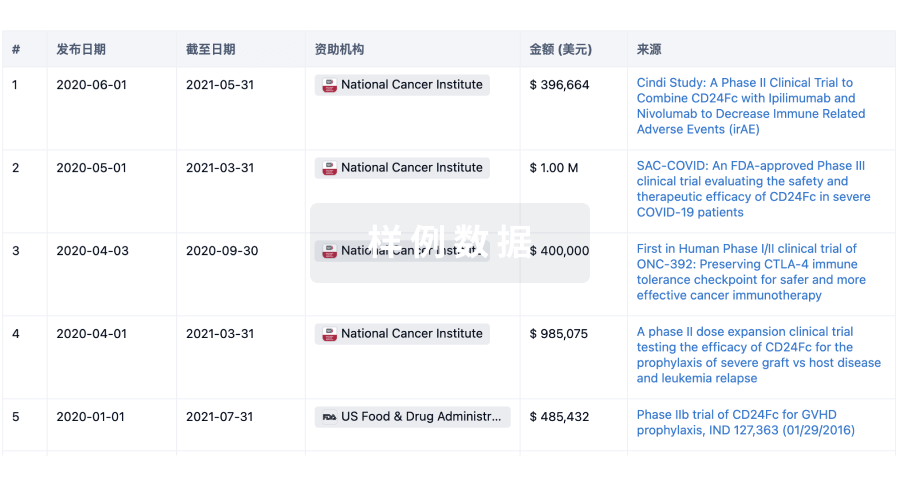
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用