更新于:2024-05-01
Nice Corp. (Japan)
更新于:2024-05-01
概览
关联
100 项与 Nice Corp. (Japan) 相关的临床结果
登录后查看更多信息
0 项与 Nice Corp. (Japan) 相关的专利(医药)
登录后查看更多信息
21
项与 Nice Corp. (Japan) 相关的新闻(医药)2024-03-08
近日,在英国国家卫生与临床优化研究所(NICE)发布的HER2低表达乳腺癌最终指南草案中,拒绝推荐使用阿斯利康/第一三共的HER2-ADC药物Enhertu。NICE表示,虽然与化疗相比,Enhertu显著改善这类患者的无进展生存期和总生存期,但是考虑到其价格,其认为不具备成本效益。NICE的拒绝促使英国乳腺癌患者组织在网上发起了一项紧急请愿,呼吁NICE、第一三共/阿斯利康尽快找到解决办法,以使得Enhertu被纳入英国国家医疗服务体系(NHS)。价格谈判未能达成共识Enhertu于2021年2月首次在英国获得批准,在该国的标价约为1455英镑(100mg/每瓶)。相比于其他治疗方案,NICE认为其不太具有成本效益。自去年12月以来,NHS也一直在与该公司就该药进行价格谈判,但双方没能达成共识。事实上,去年9月,NICE发布的指南草案征询文件中就表示了拒绝推荐Enhertu用于治疗不可切除或转移性HER2低表达乳腺癌。NICE虽然认可了Enhertu在这一群体的临床获益,但其成本效益高于到NHS所能接受的范围。不支持HER2-Low被独立分型一直以来,HER2乳腺癌的诊疗以“非阳即阴”进行二元划分,HER2过表达(免疫组化3+)或扩增(原位杂交阳性)的判定为HER2阳性乳腺癌,无上述改变为HER2阴性乳腺癌。HER2靶向药的出现大幅改善了HER2阳性乳腺癌患者的预后。不这类药物对于HER2低表达乳腺癌患者的疗效甚微,因而在临床指南中,HER2低表达乳腺癌被归为HER2阴性乳腺癌一类,采用HER2阴性乳腺癌的诊疗标准。然而,HER2阴性乳腺癌诊疗方案对这类人群的治疗效果同样差强人意,且大量相关研究发现,HER2低表达乳腺癌在生物学特征、对治疗药物的响应以及预后等方面与HER2纯阴性乳腺癌的表现截然不同。2022年6月,首个针对HER2低表达乳腺癌的Ⅲ期临床DESTINY-Breast04取得阳性结果,与化疗相比,Enhertu显著改善这类患者的的无进展生存期和总生存期。Enhertu也随后被FDA批准,成为首个针对HER2低表达乳腺癌的靶向治疗药物。随之而来的是“HER2低表达乳腺癌”被划分为一种独立HER2乳腺癌亚型”概念的风行。不过对于HER2低表达乳腺癌是否应该被独立分型仍然有较大争议。NICE委员会认为,HER2低表达仅是既往HER2阴性乳腺癌的一个亚型,应按HER2阴性方案进行诊疗。ASCO/CAP乳腺癌HER2检测指南2023年版ASCO/CAP的HER2检测更新指南中,继续沿用了2018年HER2检测推荐,即“非阳即阴”的二分法,并不推荐在HER2检测报告中引入”HER2-Low“这一报告术语。ASCO/CAP指南指出,现有的研究证据不足以支持修改HER2分型结论,建立新的HER2乳腺癌分型(如HER2低表达、HER2超低表达)还为时尚早。ESMO HER2-low乳腺癌诊疗共识此外,2023年ESMO HER2低表达乳腺癌诊疗共识中,虽然对HER2低表达、超低表达和无表达做出了定义,但并不提倡在病理报告中使用这些定义。专家共识指出,HER2低表达和HER2零表达肿瘤之间没有明显的分子特征差异。HER2低表达达不应被视为一个独立的分子亚型,而是应被视为一组异质性的肿瘤,其生物学特征主要取决于HR(激素受体)的表达水平。病理医师在报告HER2检测结果时,应保持与2018年ASCO/CAP的HER2检测指南一致的命名。在预后方面,相关研究也并未发现HER2低表达与HER2-零表达乳腺癌之间存在显著的生存差异。总结虽然目前HER2低表达乳腺癌是否可以被应被视为一种独立的HER2乳腺癌亚型仍存在一定争议,但Enhertu为该类患者群体带来的临床获益并未被否定,Enhertu作为HER2低表达乳腺癌的治疗方案已经获得美国临床肿瘤学会(ASCO)、美国国立综合癌症网络(NCCN)、欧洲肿瘤内科学会(ESMO)等指南的推荐。参考出处:https://www.fiercepharma.com/pharma/nice-poised-reject-astrazeneca-daiichis-enhertu-her2-low-breast-cancerhttps://www.biospace.com/article/uk-s-nice-rejects-astrazeneca-daiichi-sankyo-s-enhertu-for-nhs-use/英国NICE对HER2低表达的推荐可能有别于其他指南Size matters!再谈乳腺癌HER2低表达治疗概念
ASCO会议临床结果抗体药物偶联物临床3期
2024-02-27
·药研
摘 要:孤儿药的定价和报销关系到患者用药可及,对罕见病的防治与保障具有重要意义。欧洲国家在孤儿药医保准入过程中形成了卫生技术评估的特殊标准和路径,并建立了专项支付基金和多样化的风险分担协议,有效提升了孤儿药的可及性。基于此,文章选取欧洲典型国家,对比分析其孤儿药定价和报销的主要特点和共性保障思路,提出“构建孤儿药卫生技术评估加速程序、探索医保外孤儿药的分类保障机制、通过孤儿药定价和调整实现供给激励和研发激励”的建议,以期为优化我国孤儿药市场准入机制提供参考依据。孤儿药具有目标市场有限、研发成本高的特点[1],企业研发动力不足,仅靠市场机制调节并不能满足患者的用药需求,需要政府利用价格干预、定价激励等方式,保障孤儿药的可获得性。另一方面,价格高昂的孤儿药如果不纳入报销范围,将给患者带来较大的治疗负担;但纳入报销又将给医保基金带来较大的冲击,转化为重大的预算影响[2]。由此,从平衡孤儿药定价激励和医保控费的视角出发,优化孤儿药的定价和报销机制对提高我国罕见病患者用药可及意义重大。在欧盟,孤儿药虽通过EMA 集中程序注册上市,但“定价和报销”是各成员国的责任[3]。因此,欧洲各国在孤儿药的定价和报销中实践经验丰富,对我国具有一定的借鉴意义。欧洲国家大都已建立特殊的卫生技术评估(health technolog yassessment,HTA)标准,并形成了高值孤儿药报销的特别方案;荷兰、比利时、奥地利和爱尔兰等地还建立了“BeNeLuxA(Ir)”联盟,通过互认HTA 结果,以联盟谈判的方式进行孤儿药的合作定价和采购。同时,部分国家通过多样化的风险分担协议,平衡药品风险和早期获益,缩短了患者获取药物的等待时间。1 欧洲国家孤儿药的定价和医保准入机制1.1 英国的定价和医保准入机制英国先由制药公司自由定价,之后政府将通过药品价格监管计划和NICE 卫生评估的结果对价格进行调整,形成最终定价[4]。新药准入则是在接受HTA 之后,经NICE 推荐纳入到英国国民健康保障体系(Nationa Health Service,NHS)获得全额报销[5]。药品进口注册流程、申报资料撰写与上市后维护高级研讨班在 HTA 过程中,针对孤儿药,NICE 往往会接受更低的证据水平和更高的增量成本效益比(Incrementally Cost-Effectiveness Ratio,ICER)阈值[6]。对于ICER仍然超出阈值的孤儿药,NICE 将从孤儿药的社会收益、创新性、疾病患病人数、疾病的特殊性等多方面进行综合考虑,提高孤儿药纳入报销的可能性。此外,英国针对治疗超罕见疾病(患病率低于1/50 000)的孤儿药建立了高度专业化技术评估方案,其评估委员会由国家医疗服务体系人员、学术界、制药和医疗器械行业代表,患者代表、其他卫生专业人员等构成,引入了更多的价值评估维度。1.2 法国的定价和医保准入机制法国由国家卫生委员会(Haute Autorité de la Santé,HAS)下设的透明委员会(Transparency Committee, TC)承担新药的卫生技术评估工作,主要评估实际临床效益(Service Médical Rendu,SMR)和附加临床效益(Amélioration du ServiceMédical Rendu,ASMR),并分别决定报销比例和定价方式(如图1)。图1 法国孤儿药的卫生技术评估及定价报销程序从评价结果看,根据欧盟有关罕见病的认定标准,罕见病属于严重疾病[7],所以孤儿药的SMR 评估结果都较为积极,且不再要求企业提交经济证据,报销比例多在35%以上[8]。同时,由于罕见病目标适应症下治疗方案较少,孤儿药均能够获得ASMRI~III 级评价,不必受到量价协议和同类药品价格的限制,获得较高的定价水平。HAS 还特别规定,如果孤儿药目标适应症的预算影响小于每年3 000 万欧元,则直接获得ASMR I 级评级。从评估过程看,HAT 明确孤儿药根据疾病严重程度和临床急需性,可以豁免成本效益评估。且对属于创新先驱(priori innovative)的孤儿药,还可以通过加速HTA 程序,在新药上市前更早地开始HTA 评估,提前收集卫生技术评估的相关信息,从而加快HAS 的决策过程。1.3 荷兰的定价和医保准入机制荷兰主要通过“外部参考定价”的方式进行定价,即参考欧盟其他成员国的价格确定最终定价。此外,荷兰还通过“BeNeLuxA(Ir)”联盟进行谈判定价[9]。联盟主要通过互认卫生技术评估结果,提高药品定价和报销的效率,以更为透明的机制进行合作定价;通过联合价格谈判和市场调查,提高支付方在市场上的地位。对于孤儿药,BeNeLuxA(Ir)正在探索以几个国家联合谈判的方式“团购”孤儿药,并进一步降低药价。在HTA 过程中,荷兰纳入了多个利益相关者的评价维度,且十分关注新药的社会性。对于孤儿药,由于其具有更加明显的社会价值,荷兰依据医疗保险委员会(CVZ)会直接免除成本效益评估,并依据附加效益、疾病罕见性、严重程度等指标在给予其更为积极的HTA 结果。在医保准入上,CVZ 提供的HTA 报告和报销建议形成最终的报销决定。由于孤儿药往往具有临床比较优势,多数都能够获得全额报销。对于部分临床价值证据不完整、成本效益不确切或预算影响不满足要求的孤儿药,也可以申请纳入高值/特殊报销清单获得早期报销。但是,列入这类清单的药物需要提交预后成本预测和研究计划,在报销期限内持续收集临床证据,以方便后续评估[10]。1.4 欧洲国家孤儿药定价和医保准入的特点总结1.4.1 欧洲孤儿药定价和报销的特点。(1)孤儿药定价和报销依赖HTA 结果英国、法国、荷兰都通过HTA 来为定价和报销提供决策依据,并设立有专门的评估机构和专门的评估标准。HTA 的评级将在很大程度上影响孤儿药的价格水平和定价方式,并决定实际报销比例。(2)给予孤儿药科学的溢价空间。虽然每个国家HTA 立足的方法基础和价值确定方式不同,但均关注孤儿药相较现有疗法(产品)的附加效益和临床必需性。所以经过欧盟认定的孤儿药均能够获得积极的评价,从而能够以外部参考价的定价方式进入市场,给予了孤儿药很大的溢价空间。(3)以参考定价为主,探索联盟采购机制。在定价过程中,欧洲国家多采取“参考定价”和“谈判定价”相结合的方式,以“参考定价”确定药品的限价基准,然后结合HTA 评价结果和制药企业的自主报价来进行协商,确定最终药价(表1)。药品注册经理能力全面提升与实践案例分享(注册必备)对于荷兰、比利时等议价能力有限的国家,已经在逐步探索通过借助联合价格谈判和市场调查的方式, 提升支付方的议价地位,呈现“团购”趋势。1.4.2 欧洲国家孤儿药HTA 的特点。(1)建立孤儿药的特殊评估标准或路径。在评估标准上,由于孤儿药价格高昂且临床数据不足[11],各国都针对孤儿药建立了特殊的评估标准,包括提高增量成本效益阈值、增加多维度评价标准(如纳入患者及家属视角)、降低临床数据要求(如法国在效益评级过程中直接认可孤儿药的附加效益,简化数据提交)等。此外,部分国家还形成了特殊的卫生技术评估路径,如英国采取高度专业化技术评估方式评估超罕见疾病用药;法国对孤儿药设置了加速HTA 程序,在上市前即开展评估,加速孤儿药上市后的市场准入。(2)联盟采购关注HTA 的透明度和清晰度。BeNeLuxA 联盟的目标是通过合作HTA,达成联合定价和报销。目前BeNeLuxA 联盟主要确定了四种合作方式(表2)。这些合作方式均建立在统一的 HTA 标准之上,对孤儿药HTA 评价的透明度和清晰度有较高的要求[12]。而合作谈判既节省审评资源,又提高高值孤儿药的可获得性,具有一定的实践意义。2018 年,比利时将荷兰对诺西那生钠注射液的HTA 报告用于报销决策中,并通过联合谈判降价纳入到了BeNeLuxA 联合采购系统当中,实现了高值孤儿药的降价准入[13]。(3)降低孤儿药HTA 中成本效益分析要求。从评估内容看,各国均涉及附加效益评估和预算影响分析,但对成本效益的分析要求有所降低(表3)。由于各国罕见病自然史研究薄弱,孤儿药在上市后的疗效评价、成本效益分析和增量成本效益计算中都较为困难,早期成本-效益数据明显不足。因此,在评估中,基于疾病严重程度、临床急需性等标准,法国和荷兰在评估中豁免了成本-效益分析的要求。2 欧洲国家孤儿药的特殊报销策略2.1 英国孤儿药的特殊报销策略对于被NICE 拒绝推荐或者仍然处于评估状态的孤儿药,英国设立有特殊的资助项目进行补充保障,如个人基金请求、肿瘤药物基金、专项购买服务和对缺乏临床数据项目的评估购买计划。同时,英国建立了患者准入计划(patient accessschemes,PAS)和管理准入协议(Managed access agreements,MAAs)进行孤儿药的支付。PAS 是孤儿药最常用的创新支付方式,旨在提高成本效益,使患者能够获得高值孤儿药[14,15]。MAAs 则是一种有条件报销的协议。协议签订时,NHS 将设定具体的报销条件(如覆盖人群、结局指标和折扣价格等)、协议持续时间、数据收集方法和报告频率。期间企业将以商定的折扣价格提供药品,并持续收集真实世界的数据,以加强药物的证据体系,获得长期报销。2.2 法国孤儿药的特殊报销策略对于基本医保覆盖之外的孤儿药,法国还采取了两项特殊报销策略。其一是长期疾病清单(Affection Longue Durée,ALD),因严重程度或慢性性质,需要长期护理和高成本治疗的疾病将被纳入清单中,获得全额报销[16]。罕见病多为慢性病、严重疾病,往往可以被列入ALD 当中,对应的孤儿药也可获得全额报销。其二,法国建立了临时使用授权(Authorisations for Temporary Use,ATU)计划,相当于同情用药制度,旨在为罕见病患者提供尚处于临床试验阶段的孤儿药。在ATU 计划内的孤儿药批准上市后,将以post-ATU 的方式直接获得报销,直到最终决定纳入报销范围并确定最终价格,缩短了孤儿药纳入报销的时间[17]。2.3 荷兰孤儿药的特殊报销策略对于未纳入报销的高值孤儿药,荷兰引入了基于证据发展的报销(coveragewith evidence development,CED)协议[18]。政府通过与制药企业签订协议,降低对孤儿药早期临床数据和关键证据的要求,使得孤儿药可以更早得到偿付。企业则需要在协议期内,持续开展研究和证据收集工作,以验证药品带来的健康产出。协议到期后,政府根据对上市后数据收集结果确定其健康效益,而后形成最终报销决策。2.4 欧洲国家报销策略的特点总结2.4.1 重视发挥风险分担协议的作用。为保障罕见病患者用药的可及性,欧洲国家通过签订多样化的风险分担协议(Risk-Sharing Agreements,RSA)的方式,有效缩短了患者获得药品等待时间(表4)。总体来看,3 个国家涉及的风险分担协议主要有两种类型,一种是基于财务预算的管理协议,比如英国的MAAs、法国的post-ATU 报销。该类协议一般会设定成本上限或数量上限,在限定时间内规定每位患者的最大累计治疗成本或治疗计量,超过此阈值,药品制造商以折扣价格或免费提供其药物,也可能需要向支付方退款。此类协议容易操作,且管理的要求较低,应用更为广泛[19];另一种是基于健康产出结果的风险分担协议,比如英国的PAS 和荷兰的CED。这类协议则主要关注药物预期的临床效果,在某个时间节点或健康结局如果达不到承诺的效果指标,制药企业需要向支付方退款。2.4.2 构建高值孤儿药特殊支付清单。欧洲国家通过设立特殊的报销清单的方式(表5),为没有纳入医保报销范围的高值孤儿药开辟了新的报销路径。这类清单或专项基金的筛选标准可能是以“疾病”为依据,关注疾病的性质(难治性、慢性)、疾病严重程度、治疗成本等等;也可能以“药品”为依据,关注药品当前的疗效证据、定价潜力、临床急需性等等。2.4.3 关注孤儿药上市后数据收集。无论是孤儿药的管理准入协议还是有条件报销路径,都采用了“临时报销” 的共同思路,要求企业在上市后持续收集临床使用证据和真实世界数据。并通过再评估门槛的设置,提升了企业数据收集和分析的主动性。3 启示和建议3.1 构建孤儿药准入评估体系,建立加速HTA 程序建议在医保准入过程中,构建孤儿药配套的卫生技术评估体系。一方面,孤儿药临床试验招募困难,要构建完整的数据链也存在客观障碍。所以,应在评估中降低对部分数据的要求,更关注其附加临床价值。另一方面,罕见病作为一个社会性问题,应将社会效益、公平性原则等标准纳入考虑之中,减少对成本-效益分析的依赖性,构建多维度的评估体系。同时,建议我国针对孤儿药建立加速 HTA 程序。构建评估专家团队早期介入到III期临床试验中,在孤儿药上市前按照HTA 要求收集评估所需要的数据及材料,加速孤儿药在上市后的医保准入。3.2 探索孤儿药“分类”保障机制,提升药品可及为平衡医保基金影响及患者用药需求,对于尚不满足医保纳入条件的孤儿药,建议分类进行支付保障。3.2.1 建立高值孤儿药报销清单,拓宽资金筹措渠道。对于高值孤儿药建议成立专项支付基金予以报销,通过财政拨款、社会捐赠等多种方式募集基金,发挥多层次医疗保障的协同支持作用。从管理上,我国目前尚未形成孤儿药认定程序,故可针对高值的孤儿药采取“清单”管理的方式。通过设立一定的纳入标准,如疾病严重程度、替代疗法情况、临床急需程度等等。优先保障罕见病目录中纳入、且无替代治疗方法的创新孤儿药,做好罕见病目录与孤儿药报销清单的有效衔接。3.2.2 丰富风险分担协议类型,促进上市后数据收集。对于临床证据不足的孤儿药可引入风险分担协议,对上市前期的风险进行控制。建议在我国在前期探索经验的基础上,对于早期临床效果数据不充分的创新孤儿药,引入基于临床效果指标的风险分担协议,以促进长期的临床证据收集和罕见病自然病史的研究。并不断丰富风险分担协议类型,规范协议的制定、执行及管理流程,扩展应用范围。3.3 巧用溢价激励,配合价格调整机制中国作为亚洲国家定价的参考国之一,过低的溢价比例将会影响制药厂商进入中国市场和保障供应的积极性。因此,建议我国在谈判和采购过程中,选取合理的采购方式,给予孤儿药一定溢价比例,激励本土研发和引进。对于一些不具有临床替代性的廉价孤儿药,因利润有限出现短缺的,也可以采取反向的调节机制,允许孤儿药根据临床疗效评价结果和该年的销售情况获得一定的成本补偿。4 结语罕见病作为一个国际性的公共卫生问题,解决好孤儿药的可及问题具有较高的社会价值。因此,为鼓励罕见病领域的药品创新,我国仍然需要保持溢价激励和价格调节机制,但为平衡医保控费,保障药品可及,还需进一步优化孤儿药的评估和支付方式。在卫生技术评估中应充分体现孤儿药的临床价值及社会价值,做好孤儿药上市后疗效数据和安全性数据的收集。在孤儿药的支付过程中,有效发挥风险分担协议、高值孤儿药基金的补充支付作用,优化多层次医疗保障体系。------------THE END------------内容来源:《中国卫生经济》点击查看:投稿获丰厚稿费关注药研 一路同行药研论坛:始终以为药品研发一线人员提供高质量、高性价比培训为第一宗旨。自成立以来,累计举办80余期药品研发、注册领域研讨班。据统计,中国医药工业百强企业和研发百强企业均超过90%参加过药研收费类培训。截至2023年底,包括上海强生、辉瑞、阿斯利康、山德士、日本大冢、大鹏、卫材、小林制药、扬子江、恒瑞、正大天晴、东阳光、科伦、中科院等在内的2800余家企事业单位参加过药研线下收费培训,得到业界普遍认可与好评!药研学院:已同众多企业合作超百期直播课,全网观看突破100万+,药研直播课聚焦药品研发相关主题,平均观看人数行业领先。药研自媒体矩阵20万+:目前药研公众号研发领域关注用户约10万人!双直播平台10万人,微信社群5万人,其中制药企业和研发机构关注量5000+。商务合作:15911172616
孤儿药
2023-04-14
·生物谷
减肥是绝大多数人的“人生必修课”,更有甚者每年都要修一次。 (尝试)减过肥的人都知道,“管住嘴迈开腿”并不是件容易的事儿,减肥路上总有很多痛苦与阻碍,这也是为什么人们致力于寻找到“两全”的减肥法。在度假照被嘲笑肥胖后,特斯拉CEO Elon Musk于去年在社交媒体上晒出自己“暴瘦”的图片——仅一个月的时间就成功减去20磅(约9kg)!而他自曝成功减肥的秘诀是:禁食与Wegovy(活性成分:司美格鲁肽 Semaglutide)。马斯克减肥前后对比图司美格鲁肽,何许减肥利器也?事实上,司美格鲁肽最早是作为“一周打一次”降糖药而为人熟知。司美格鲁肽是一款新型长效胰高糖素样肽-1(GLP-1)类似物,可以有效抑制胰高血糖素的释放,从而达到降低血糖的作用,于2021年4月在中国上市。因其体内半衰期在1周左右,实现每周给药一次,迅速取代了上一代降糖药。就在上个月,司美格鲁肽再度传来好消息:口服版III期临床试验数据出炉,高效减糖减重,甚至无需注射!口服司美格鲁肽的III期临床数据2023年3月24日,口服版司美格鲁肽(25mg和50mg)治疗2型糖尿病成人患者的IIIb期PIONEER PLUS研究的数据公布,受到了极为广泛的关注。与此前获批的14mg剂量相比,25mg和50mg剂量的司美格鲁肽更大程度上降低了糖化血红蛋白(HbA1c),从治疗策略角度来看分别下降了1.8%和2.0%,高于14mg的1.5%。当然,除了有效降低血糖之外,另一个“意外”的效果引起了研究者的关注——减重效果相当惊人。具体来说,每日一次口服14mg、25mg、50mg的司美格鲁肽,52周后体重能分别减轻4.4kg、6.7kg和8.0kg。司美格鲁肽的减肥效果使其又火了一把,甚至被冠上“减肥神药”之称!其实,司美格鲁肽控制体重的作用算是“顺带”,因其能够模拟胰高血糖素的作用,增加了人体对胰岛素的敏感性。在此基础上,饭后的饱腹感提升了,胃排空的速率减慢了,自然能够减少饥饿感、热量摄入以及体重。当然,上个月最新公布的口服版司美格鲁肽III期临床试验数据是基于2型糖尿病患者的,在非糖尿病患者中也会有同样的减重效果吗?早在2021年3月,医学顶刊之一《NEJM》发布的全球性重磅临床试验显示:仅需每周给药一次2.4mg,司美格鲁肽使得肥胖参与者的体重狠狠地下降了15.3kg!在整个试验期间,有将近七成的参与者体重减轻了10%以上,而降低15%以上的人数超一半。DOI: 10.1056/NEJMoa2032183研究者从全球16个国家开展了随机、双盲、安慰剂对照试验,最终纳入了1961名BMI≥30(或BMI≥27但合并有≥1种体重相关疾病,比如高血压、血脂异常、阻塞性睡眠呼吸暂停或心血管疾病)的成年人,但所有参与者均没有糖尿病。按照2:1的比例,参与者被随机分配到试验组或安慰剂组。其中,试验组接受司美格鲁肽治疗,每周皮下注射一次,每次剂量为2.4mg,为期68周;而对照组则接受安慰剂。此外,全员都会接受生活方式干预。结果显示,司美格鲁肽起效快且效果好。在使用司美格鲁肽的第4周左右,就能观察到参与者的体重下降;而到第60周时,体重会降至最低点。在整个持续68周的试验期间,司美格鲁肽组的体重平均减少了14.9%,明显差别于安慰剂组的2.4%。具体来说,从基线到第68周,司美格鲁肽组的平均体重减少了15.23kg,显著高于安慰剂组的2.6kg。相比于基线体重,接受司美格鲁肽治疗的参与者中有86.4%的体重减少了超5%,69.1%的体重减少超10%,更有50.5%的“狠人”体重减少了超15%!试验组与对照组的体重变化情况事实上,除了体重以外,司美格鲁肽组参与者的总脂肪量以及内脏脂肪均显著减少,腰围、BMI、血压、血糖等指标也出现喜人的变化!那么,司美格鲁肽的安全性如何呢?相信这也是不少人使用减肥药的犹豫之处。有趣的是,司美格鲁肽组和安慰剂组中报告不良事件的比例相似。司美格鲁肽组中最常报告的副作用为胃肠道疾病,比如恶心、腹泻、呕吐和便秘等,约占74.2%;但大多数情况并不严重,多为轻至中度,且是一过性的。不过值得注意的是,约有9.8%使用司美格鲁肽治疗的参与者出现了严重的不良反应,包括胃肠道疾病和肝胆疾病。综上,本篇研究显示,司美格鲁肽的减重效果还是相当不错的,尤其是对于肥胖群体。毕竟谁不想“无痛且高效”地减肥呢?2021年6月,FDA正式批准了司美格鲁肽(商品名:Wegovy)用于治疗单纯肥胖的这个适应症,且写在了说明书上。至此,司美格鲁肽的减肥效果得到了“官方认证”。看到这儿,相信不少“以减肥为终身事业”的读者内心开始蠢蠢欲动了。但接下来,小编要来泼一盆冷水。首先,现阶段“减肥神药”司美格鲁肽在我国获批的适应症仅为2型糖尿病,并未获批减肥适应症。同时,目前我国上市的司美格鲁肽并非上文提及的Wegovy,而是Ozempic,该药甚至都未获得FDA的批准。不过,好消息是,就在4月6日,口服版司美格鲁肽(Wegovy)的III期临床试验将在国内启动,相信很快就能有国人数据出来。其次,即使在国内获批减肥适应症,司美格鲁肽也有固定的适用人群,而不具有普适性。正如上文研究中提到的那样,该药适用于BMI≥30的患者,或BMI≥27但合并有≥1种体重相关疾病的患者。这条适应症也更新在药物说明书中。司美格鲁肽的适应症(来源:FDA)正如NICE药品评估主任Helen Knight强调的那样,该药物并不是每个人都可以长期使用的。我们已经提出了具体的使用建议,以确保药效。当然,司美格鲁肽也不是“一打就瘦的减肥神药”,而需要配合饮食以及生活方式的管理。而在停药之后,很容易出现反弹的现象,据诺和诺德高级副总裁Karin Conde-Knape所述,“现有数据表明,大多数人在停止使用Wegovy后的五年内会恢复大部分体重,并在两到三年后恢复大约50%的体重。”所以,司美格鲁肽究竟是不是“胖子救星”?让我们拭目以待吧。参考文献:Khoo TK, Lin J. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N Engl J Med. 2021;385(1):e4. doi:10.1056/NEJMc2106918 撰文 | Swagpp编辑 | Swagpp来源 | 梅斯医学会议推荐点精彩推荐:1、蛋糕烧烤自由实现了?Nature子刊:抵御高脂饮食不利影响的方法找到了,或将不再是健康的威胁!2、扎心!收入少死的早?两篇JAMA研究超百万数据显示:低收入者比高收入同龄人的死亡率高10%至20%!3、首次!德国Alessandra Moretti教授研究13年后,这颗“迷你心脏”类器官终于拥有了完整的“外衣”!4、闻闻味道就能瘦?iScience:伴侣独特的气味不但助眠抗衰,还能减重、延长寿命8%!5、小酌有益健康?不存在的!JAMA子刊超480万人研究:女性每日饮酒超25克、男性每日饮酒超45克全因死亡风险显著增加!
临床3期临床结果临床成功
100 项与 Nice Corp. (Japan) 相关的药物交易
登录后查看更多信息
100 项与 Nice Corp. (Japan) 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年07月08日管线快照
无数据报导
登录后保持更新
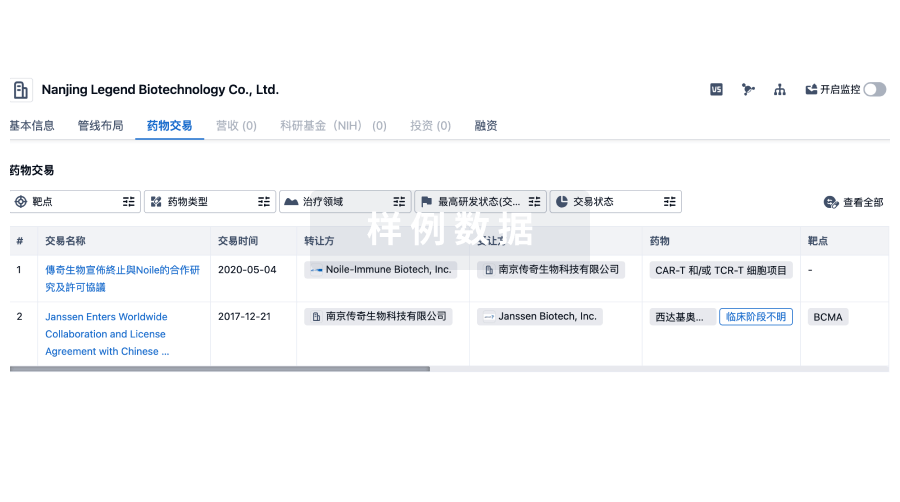
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
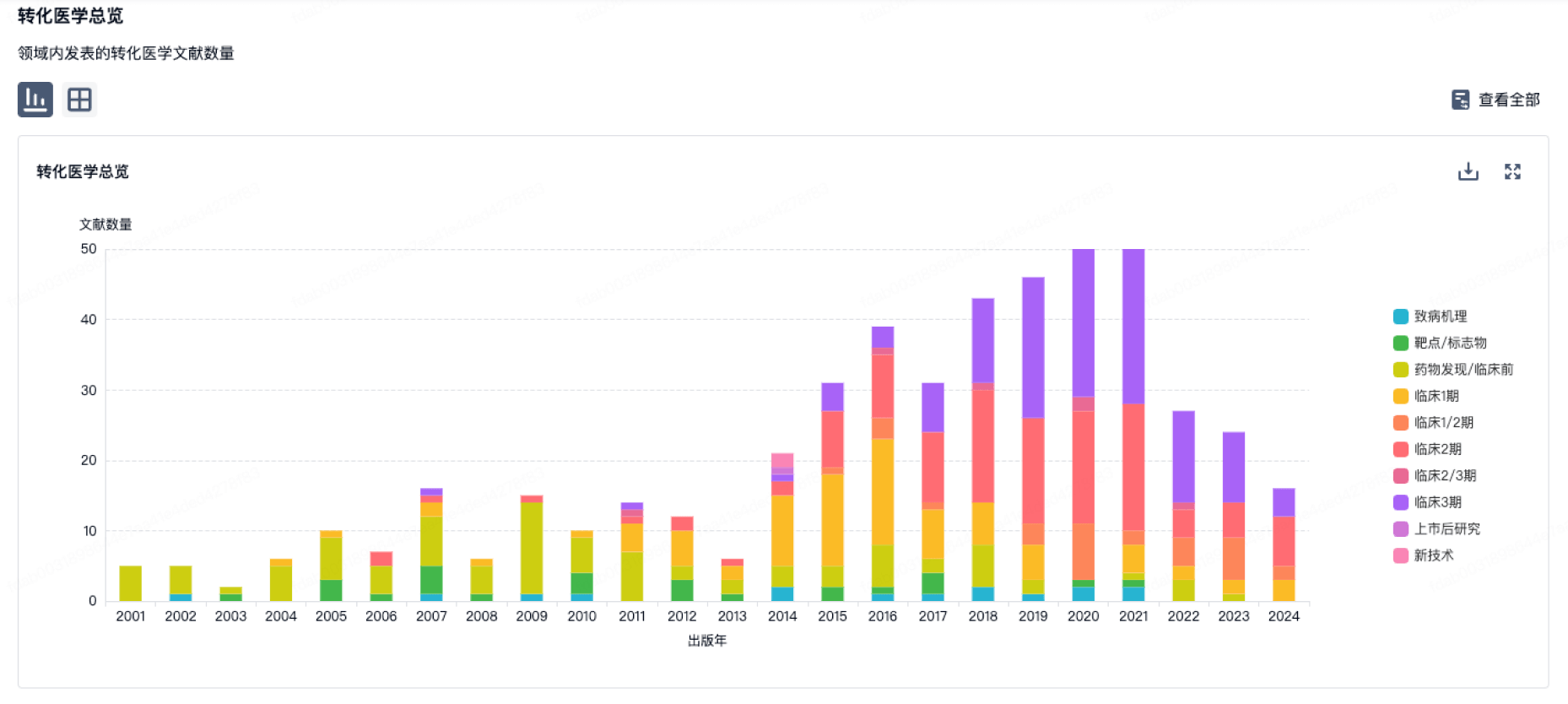
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
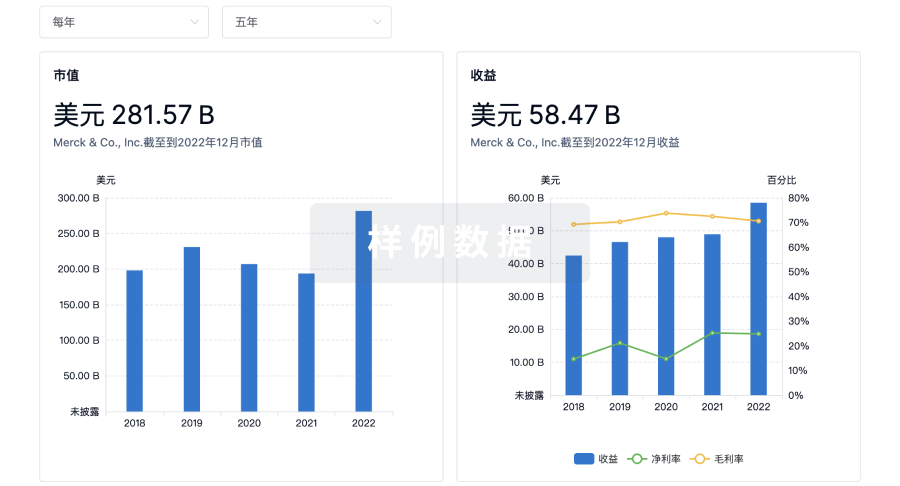
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
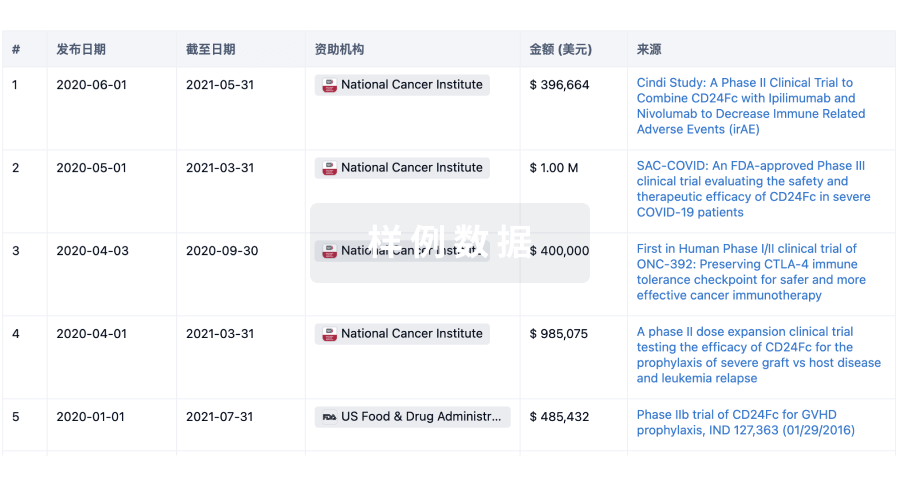
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用