预约演示
更新于:2025-05-07
Triarm Therapeutics Ltd.
更新于:2025-05-07
概览
标签
肿瘤
血液及淋巴系统疾病
免疫系统疾病
CAR-T
细胞疗法
TCR-T细胞疗法
疾病领域得分
一眼洞穿机构专注的疾病领域

暂无数据
技术平台
公司药物应用最多的技术
暂无数据
靶点
公司最常开发的靶点
暂无数据
| 排名前五的药物类型 | 数量 |
|---|---|
| CAR-T | 11 |
| 细胞疗法 | 3 |
| 自体CAR-T | 1 |
| TCR-T细胞疗法 | 1 |
关联
17
项与 星尘生物科技(上海)有限公司 相关的药物作用机制 CD19抑制剂 [+1] |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症- |
最高研发阶段临床1期 |
首次获批国家/地区- |
首次获批日期- |
靶点- |
作用机制- |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症- |
最高研发阶段早期临床1期 |
首次获批国家/地区- |
首次获批日期- |
靶点- |
作用机制- |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症- |
最高研发阶段早期临床1期 |
首次获批国家/地区- |
首次获批日期- |
100 项与 星尘生物科技(上海)有限公司 相关的临床结果
登录后查看更多信息
0 项与 星尘生物科技(上海)有限公司 相关的专利(医药)
登录后查看更多信息
54
项与 星尘生物科技(上海)有限公司 相关的新闻(医药)2024-09-13
Industry veteran
Doug Williams
is back with a new approach to CAR-T cell therapies that he expects to take into the clinic in the US by year’s end.
After a
brief stint
at
Sana
Biotechnology
,
leaving his R&D chief post
in the spring, Williams is returning to the role of CEO at a little-known biotech. In recent weeks, he took the top spot at China-born and now
California-headquartered biotech
TriArm Therapeutics
.
It’s a position he held at
Codiak BioSciences
, a startup that tried turning exosomes into medicines, but the relatively nascent idea
couldn’t survive
on the public markets in 2023.
Now, he’s driving TriArm’s mission to “democratize” CAR-T cell therapies using what it calls a “fast-in-time” (FIT) and non-viral manufacturing process, Williams told
Endpoints News
. He said TriArm’s technology platform has led to a two-day manufacturing process.
“There’s also some potential advantages to cell fitness, the actual fitness of the CAR, the composition of cells that we’re reinfusing back into the patient,” Williams said.
“We’re preserving a population of cells using this FIT process that may end up being more fit from a biologic perspective because they’re enriched in STEM and naïve T cells,” he added.
TriArm has been relatively quiet since launching in 2019. Its financial backers include
Panacea
Venture
,
MSD
Capital
and
Lombard
Odier
, and it employs about 100 people across Shanghai, Taipei and San Mateo, CA.
The company aims to speed up the testing of CAR-T therapies and make faster go/no-go R&D decisions by employing an investigator-initiated model in China and then moving into clinical studies in the US, Williams said.
“As somebody that is a drug developer by choice and by training, to be able to get a glimpse of what is arguably real data in a population of patients as sort of a preamble to filing an IND and really moving ahead in larger, much more expensive trials, it’s a fantastic setup to have available,” Williams said.
Its first US-based trial, testing a CD19-directed autologous therapy, is slated to start later this year, Williams said. A gene therapy could soon enter a similar patient setting with the
emergence this week
of a new biotech called
Vironexis
.
—
Kyle LaHucik
→
Biogen
’s board continues to evolve under chair
Caroline Dorsa
, who
replaced
Stelios Papadopoulos
last year. On Jan. 1, ex-
AstraZeneca
BioPharmaceuticals R&D chief
Mene Pangalos
will take a seat
on the board, and
Lloyd Minor
— the dean of Stanford University’s School of Medicine — will become a board member much earlier on Oct. 1. After the hubbub surrounding the
departure
of board member
Alex Denner
and the
appointment
of his romantic partner
Susan Langer
in June 2023, Biogen then elected ex-
3M
president and CFO
Monish Patolawala
to the board in November.
Richard Mulligan
and
William Jones
also left the board last year. Pangalos
retired
from AstraZeneca in April and
was replaced
by
Sharon Barr
.
→
Vir Biotechnology
’s revamp
took the spotlight
in early August, as CEO
Marianne De Backer
made the decision to prop up its hepatitis B and D development and wind down its Covid-19, flu and T cell-based viral vector programs. On Wednesday, Vir said that
Jason O’Byrne
will succeed
Sung Lee
as CFO on Oct. 2. O’Byrne had the same title at
Caribou Biosciences
since February 2021 and is the former SVP of finance at
Audentes Therapeutics
. He spent 13 years with
Genentech
in a number of roles, including head of commercial finance for the Asia-Pacific region. To accommodate the makeover, Vir laid off about a quarter of its staff.
→
BridgeBio Oncology Therapeutics
has selected
Yong Ben
as chief medical and development officer. Readers may know Ben from
BeiGene
, where he was CMO, solid tumors from 2019-22, and he’s been a venture partner at
Eight Roads
for the past two years. Ben was the global clinical leader of immuno-oncology clinical development at AstraZeneca when
Imfinzi
received its
first approval
for advanced bladder cancer in May 2017, and a week later, he became CMO of
BioAtla
. BridgeBio’s oncology spinout
debuted
in May with a $200 million infusion from
Cormorant Asset Management
,
Omega Funds
,
GV
and others.
→ As
Endpoints News
reported
on Monday,
Eli Lilly
ended a three-month search by promoting
Lucas Montarce
to CFO. He replaces current
Alphabet
finance chief
Anat Ashkenazi
, with an interim stint by controller
Gordon Brooks
sandwiched in between. Montarce’s career at Lilly began in 2001 and has held enough positions to scroll through ever since. He was elevated to president and general manager for the Spain, Portugal and Greece hub in February after nearly three years as group VP, corporate controller and chief financial officer for
Lilly Research Laboratories
. He takes over as CFO at a time when the Indianapolis pharma’s market cap has exceeded $800 billion.
→
Pfizer
CSO
Mikael Dolsten
has nabbed
a spot on the board of directors at
Rocket Pharmaceuticals
. Dolsten
announced in July
that he would leave Pfizer after 15 years, but he’s sticking around until the company finds a successor. In late June, questions about manufacturing
resulted in a CRL
for
marnetegragene autotemcel
(
marne-cel
), Rocket’s gene therapy to treat an ultra-rare disease called severe leukocyte adhesion deficiency-I.
→
Meg Alexander
has been promoted to president and COO of
Ovid Therapeutics
, which is on the verge of starting a Phase 2 study of its
Graviton
-partnered ROCK2 inhibitor in cerebral cavernous malformations. This is Alexander’s third appearance in as many years after she was named
chief strategy officer
last year and
corporate affairs chief
in 2022. “Her extensive experience, operational expertise, and business acumen position her well to lead the next phase of the company’s evolution,” Ovid CEO
Jeremy Levin
said in a statement
.
→ Sana Biotechnology finance chief
Nate Hardy
will resign “for personal reasons” on Oct. 4,
according to an SEC filing
. Hardy was on the finance team at
Juno Therapeutics
with CEO
Steve Harr
before he took the CFO job at Sana in 2018. The Seattle-based biotech
just hired
UCB
’s
Dhaval Patel
as its CSO.
→ After five years on the job,
Forge Biologics
CEO
Tim Miller
is stepping down
, making way for commercial chief
John Maslowski
to take over Oct. 1. Maslowski initially joined Forge as chief commercial officer in June 2021. Before that, he was president and CEO of
Castle Creek Biosciences
and
Fibrocell
Science
.
→
Matt Shaulis
is headed to BeiGene as general manager of North America on Sept. 25, while
Josh Neiman
— the chief commercial officer for North America and Europe — has stepped down “to pursue a new entrepreneurial opportunity at a healthcare technology startup,”
according to a release
. For the last year and a half, Shaulis has been chief commercial officer and US president for Swedish drug developer
Hansa Biopharma
, and he took on the role of president, inflammation and immunology for the international developed markets at Pfizer from 2018-21. At the end of his seven-year tenure with the pharma giant, he was elevated to SVP for global commercial and medical go-to-market model transformation.
→
Jeff Hartness
has been named
chief commercial officer at I&I biotech
Apogee Therapeutics
, which
hit the Nasdaq
with
Sagimet Biosciences
on July 14, 2023. Hartness spent more than 15 years with
Sanofi
before jumping to
Bausch Health
in 2016 as VP, strategic account management. Last year, he was promoted to EVP and head of Bausch Health’s market access, commercial operations, policy and government affairs teams, while serving as general manager of the neurology portfolio and generics businesses. Chaired by ex-
Prometheus Biosciences
chief and current
Mirador Therapeutics
CEO
Mark McKenna
, Apogee is trying to distinguish itself in the atopic dermatitis space with its monoclonal antibody
APG777
.
→ The team at non-opioid pain biotech
Latigo Biotherapeutics
is quickly taking shape under new CEO
Nima Farzan
, who
succeeded
Westlake Village BioPartners
’
Desmond Padhi
in late July. After bringing in medical chief
Neil Singla
last month, Latigo
has tapped
William Blair
analyst
Tim Lugo
as CFO. Lugo joined William Blair as a senior associate in 2005 and would eventually become a partner, leading the biotechnology equity research team. Latigo also added Westlake managing director
Beth Seidenberg
and
Foresite Capital
’s
Jim Tananbaum
to the board of directors.
→ Now that
Rytelo
(
imetelstat
) is being sold in the US for lower-risk myelodysplastic syndromes with transfusion dependent anemia,
Geron Corporation
has recruited
Jim Ziegler
as chief commercial officer. Ziegler had been EVP, commercial for
Iovance
, which secured the long-awaited
FDA approval
of its TIL therapy
Amtagvi
for melanoma in February. He also spent a decade in two separate stints with
Gilead
, culminating in his role as VP of the cardiopulmonary and inflammation business unit.
→
Alan List
is retiring
as CMO of
Precision BioSciences
, and 12-year
Merck
vet
Murray Abramson
has jumped on board to lead clinical development. Abramson left Merck in 2011 to become VP of global clinical operations at Biogen, a position he held for nine years. From 2020-22, Abramson was SVP of clinical innovation for
Tempus
. Precision also said in a release that ex-
Aligos Therapeutics
EVP
John Fry
will be a strategic clinical advisor for its hepatitis B program. Following his
resignation
as president and CEO of Moffitt Cancer Center over his connections to China, List
reemerged
as Precision’s medical chief in 2021.
→
Rami Daoud
has made his way
to gene therapy developer
Affinia Therapeutics
as chief business and financial officer. Daoud comes off a yearlong stint as president of
GeneScience Pharmaceuticals USA
and was a corporate development exec with
Amarin
.
Rick Modi
’s crew was an Endpoints 11 winner
in 2021
and had IPO aspirations that
ultimately faded
in late 2022.
→
HC Bioscience
has recruited
Simon Tsang
to replace
Christine Foster
as CBO. Tsang is an
Amgen
corporate development alum who was VP, business development for
Tesaro
from 2016-19. After
GSK
bought Tesaro for $5.1 billion, he landed at
Pivotal bioVenture Partners
as an operating partner. HC Bioscience is in a small-but-emerging group of biotechs
working on
tRNA-based drugs, along with
Flagship
’s
Alltrna
and
Tevard Biosciences
.
→
Rivus Pharmaceuticals
, which
claimed a Phase 2a win
last month with its drug for obesity-related heart failure with preserved ejection fraction,
has appointed
Tom O’Neil
as CFO and ex-
ProfoundBio
COO/CFO
Erin Lavelle
to the board of directors. As finance chief, O’Neil helped lead
Satsuma Therapeutics
to its IPO in 2019
and its sale
to
Shin Nippon Biomedical Laboratories
last year. Lavelle orchestrated
ProfoundBio
’s
$112 million Series B
in February, and
Genmab
would agree
to buy
the ADC biotech less than two months later. She also
picked up
another board seat this week at Aviceda Therapeutics.
→ We have an impressive lineup of CMO appointments: First, Eli Lilly’s obesity and metabolic disease partner
HAYA Therapeutics
has named
Jordan Shin
as CMO. Shin had been SVP of clinical development and translational science (cardiology) for gene therapy player
Lexeo Therapeutics
, which
went public
last November with a $100 million IPO. Earlier, he worked for
Renovacor
as SVP of clinical development and translational science until it was sold to
Rocket Pharmaceuticals
. Up to $1 billion is on the line with HAYA and Lilly’s recent
drug discovery pact
.
→ Canadian psychedelic biotech
Reunion Neuroscience
has brought in
Mark Pollack
as medical chief. Pollack briefly worked for
Sage
as SVP for global medical affairs and was part of a
leadership shakeup
that also involved the departures of CSO
Al Robichaud
and development chief
Jim Doherty
. Reunion went private last summer with the help of
MPM BioImpact
, and a $103 million Series A
followed in May
from MPM, Novo Holdings and several other investors.
→ A week after Reunion’s Series A financing, San Diego upstart
Aardvark Therapeutics
raised $85 million
in a Series C to bankroll late-stage trials for its Prader-Willi syndrome asset
ARD-101
. And like Reunion, Aardvark
has found
a new medical chief.
Manasi Sinha Jaiman
was promoted to the same position at stem cell therapy maker
ViaCyte
before it was sold to
Vertex
in 2022
for $320 million
, and she would stay at Vertex as a VP. In the same release, Aardvark included the appointment of regulatory affairs leader
Tom Bicsak
, who came on board in July.
→
Milan-based
AAVantgarde Bio
is picking up
Jayashree
Sahni
as CMO. Sahni is taking over the reins from
Naveed
Shams
, who had served in the role since 2022. Sahni joins the team from
Novartis
, where she was senior global program clinical lead. Earlier in her career, Sahni was with
Roche
, where she worked on the Phase 1 and 2 clinical trials for
Vabysmo
.
→ Elsewhere,
RA Capital
-backed
Tyra Biosciences
has recruited
Doug Warner
as CMO. Warner joins the California-based crew from
eFFECTOR
Therapeutics
, where he served in the same role. Previously, Warner had an 18-year stint at Amgen, where he led development of drugs like
Vectibix
,
Xgeva
and
Prolia
. Warner’s appointment comes after Tyra
picked up $200 million
in PIPE financing in February to progress its oral FGFR3-selective inhibitor,
TYRA-300
.
→
Longwood
’s
Solu Therapeutics
, which
debuted in August 2023
with a $31 million seed round,
has welcomed
Sergio Santillana
as CMO. Peer Review told you about Santillana’s
resignation
from the same position at
Ikena Oncology
in February, and he’s also the ex-CMO at
Merrimack Pharmaceuticals
and
Ariad Pharmaceuticals
. Earlier in his career, Santillana had a string of roles at Eli Lilly, GSK and
Takeda
.
→
Maud Brandely
is retiring after eight years as CMO of French cancer biotech
Transgene
, and
Sanofi Genzyme
vet
Emmanuelle Dochy
has replaced her
. Dochy just completed a seven-year run at
Bayer
, where she recently served as VP of global medical affairs for the tyrosine kinase inhibitor (TKI) oncology franchise and scientific partnerships head. Meanwhile, Transgene has also enlisted Roche oncology alum
Maurizio Ceppi
as CSO.
→
KalVista Pharmaceuticals
has named
Brian Piekos
as CFO. Far from his first run as finance chief, Piekos served in the role at
Elicio
Therapeutics
,
Gemini Therapeutics
and
AMAG
Pharmaceuticals
. In May, KalVista
announced
that it was dropping its preclinical program in hereditary angioedema in favor of focusing on its HAE pill
sebetralstat
. At the time, KalVista said it would find a partner for its dropped program.
→
Beverley Carr
has taken the CBO post
at London-based
Charm Therapeutics
, a startup co-founded by the University of Washington’s
David Baker
that uses its 3D deep learning tech DragonFold to discover new drugs. Carr is a GSK business development vet who had been chief business and operating officer for
Amphista Therapeutics
and then stepped into the role of interim CEO when
Nicki Thompson
left.
→
In July,
Cue Biopharma
announced
that it was prioritizing its autoimmune pipeline and slashing 25% of its staff. Now,
J&J
vet
Lucinda Warren
has joined
the Boston team as CBO. Warren’s 10-year stint at J&J culminated in her role as VP of business development for neuroscience and Japan regionally. Earlier in her career, Warren was with
Janssen Cilag Australia
and
Centocor
/
Janssen
Biologics
.
→ Verily
, the healthcare company spun out of
Google
parent
Alphabet
,
has rolled out
the welcome mat for
Michael Radwin
as its new head of data science and AI. Radwin joins from the CDC, where he was a founding member of the Office of Public Health Data, Surveillance, and Technology. Radwin’s résumé also includes roles at
Intuit
and
Yahoo
. Earlier in June, Verily announced that it was
jumping into
the GLP-1 business with the launch of a new virtual program to help patients manage cardiometabolic diseases with care teams and medications.
→
Dutch biotech
VarmX
, which is developing a
reversal agent
for blood thinners,
has promoted
Martijn Negen
to the role of COO. Negen joined VarmX last September as SVP of global commercial strategy & business development from
AM-Pharma
. He also has experience from
Portola
Pharmaceuticals
,
Teva Global Specialty Medicines
and Genentech.
→ Opthea
has announced
a slew of changes to its leadership team. First, the company is losing commercial chief
Judy
Robertson
, CFO
Peter Lang
and SVP, global clinical operations
Bruno
Gagnon
, effective Sept. 9. Filling in for Lang and Robertson in the interim will be
Danforth
Advisors
managing director
Daniel Geffken
and Genentech and Novartis vet
Mike
Campbell
, respectively. Meanwhile,
Jen Watts
will succeed Gagnon. That’s not all: Opthea is also picking up
Dayong
Li
as SVP, biometrics and
Anthony
Bonifazio
as VP, market access.
→ UK respiratory disease biotech
Enterprise Therapeutics
has introduced
Annabella Amatulli
as head of regulatory affairs. The
Janssen
vet was the global regulatory sciences leader for
Alfasigma
, the Italian company that bought
Ocaliva
maker
Intercept Pharmaceuticals
.
→ Tonix Pharmaceuticals
has brought aboard
Thomas Englese
as EVP of commercial operations. Englese was formerly chief commercial officer of
Tris Pharmaceuticals
and
Aziyo
Biologics
. Before that, he had an 11-year run with
Mallinckrodt
, culminating in his role as SVP and general manager of North America hospital therapies.
→ A handful of VCs have been making some moves. First up:
Abingworth
has turned to
SFJ Pharmaceuticals
founder and executive chair
Robert DeBenedetto
to tackle the role of managing director, clinical co-development;
Sofinnova Partners
has added
a new partner.
Karl Naegler
just completed a four-year run as managing partner with
Wellington Partners Life Sciences
in June; and finally,
Frazier Healthcare Partners
has named
Willis Chandler
as an executive-in-residence on the growth buyout team. Chandler left
Cencora
(formerly
AmerisourceBergen
) in June after nearly three years as president of global pharma services and six years overall.
→ Co-founded by
Jim Wilson
,
Passage Bio
has elected
Ultragenyx
CBO
Tom Kassberg
to the board of directors. One of Wilson’s new companies,
GEMMA Biotherapeutics
,
licensed
a trio of gene therapy candidates from Passage Bio in early August.
→
Enoch Kariuki
has joined
the board of directors at
Cyrus Mozayeni
’s
ADC shop
Pheon Therapeutics
, which is chaired by ex-
Blueprint Medicines
chief
Jeff Albers
. Kariuki became president of
Endeavor BioMedicines
this year and ran
Lengo Therapeutics
until
it was purchased
by Blueprint in 2021.
→
Yoshiharu Mizui
now has a seat
on the board of directors at
SEED Therapeutics
, the biotech near Philadelphia that struck a “molecular glue” deal with
Eisai
last month. Mizui is the president of Eisai’s VC arm,
Eisai Innovation
, and he also juggles two other roles as head of collaboration & incubation in the DHBL (Deep Human Biology Learning) office and head of academic & industry alliance.
→
Creyon Bio
has made room
for
DCVC Bio
operating partner
Serge Messerlian
and
Lux Capital
principal
Shaq Vayda
on the board of directors. Messerlian has board seats at
Auron Therapeutics
and
Plexium
, and Vayda’s numerous board credits include
Atavistik Bio
and
Enveda Biosciences
.
高管变更IPO上市批准细胞疗法
2024-08-21
1981年,英国剑桥大学科学家Martin Evans和同事们成功从小鼠胚胎中分离出胚胎干细胞,奠定了细胞与基因治疗的基础。此后,细胞与基因治疗(CGT)技术迅速发展。
2017年,诺华Kymriah登陆美国,成为FDA批准的第一个CAR-T细胞疗法;同年,FDA批准全球首款基因治疗产品Luxturna;2023年,基于CRISPR/Cas9基因编辑疗法Casgevy的获批,开启了基因编辑的新时代。
CGT的技术迭代和在临床上取得的成功,将越来越多的产品推向市场。IQVIA报告显示,截至2023年底,总计76款细胞和基因疗法获得全球监管机构的批准上市,与2013年的总数相比翻了一倍多。
尤其在中国,CGT已成为生物制药的一块创新热土,并催生了许多现象级产品。这一定程度刺激资本市场的活跃:截至今年上半年,国内超过40家CGT公司披露新一轮融资。
1
“全球首款”上市疗法
1
CAR-T疗法
• 首款治疗ALL和MCL的CAR-T疗法——吉利德
2020年7月,吉利德靶向CD19的自体CAR-T疗法Tecartus获FDA加速批准,用于治疗先前接受过2种或多种系统疗法(包括一种BTK抑制剂)的R/R MCL成人患者。
Tecartus是继诺华Kymriah和Kite的Yescarta之后第三个获得FDA批准的CAR-T疗法(后两者均于2017年获得批准),是第一个被批准用于治疗成人复发/难治性B细胞前体ALL患者的CAR-T细胞疗法,同时也是全球首个MCL CAR-T疗法。
财报显示,2024上半年,Tecartus营收2.07亿美元,同比增长17%。
• 首款BCMA CAR-T疗法——BMS&2seventy bio
2021年3月,全球首个BCMA CAR-T疗法Abecma获得FDA批准,用于治疗既往至少接受过4种癌症治疗无效或复发的多发性骨髓瘤患者。该产品由BMS和2seventy bio联合开发。
BCMA是一种几乎普遍在多发性骨髓瘤癌细胞上表达的蛋白质。作为一种抗BCMA CAR-T细胞疗法,Abecma识别并结合BCMA,导致表达BCMA的细胞死亡。
Abecma是获批治疗复发性/难治性多发性骨髓瘤的三种CAR-T疗法之一,也是FDA批准的第五款CAR-T疗法。2024上半年,Abecama营收1.77亿美元,同比下降37%。
• 首款获得FDA批准的CGT“国货”——传奇生物&强生
2022年2月,由强生和传奇生物共同开发的CAR-T疗法西达基奥仑赛(商品名Carvykti),获得FDA批准上市,用于治疗复发/难治性多发性骨髓瘤成人患者。这是首款由中国技术转移获得FDA批准的细胞治疗产品。
2024年4月,FDA扩大了Carvykti适应症批准范围,令其成为首个且唯一获批用于多发性骨髓瘤患者二线治疗的BCMA靶向疗法。
根据金斯瑞发布的公告,Carvykti在2024年第二季度销售达到1.86亿美元,相比第一季度1.56亿美元增长了3000万美元,半年销售额达到3.42亿美元。
2
TIL疗法
• 全球首款TIL疗法——Iovance Biotherapeutics
2024年2月,FDA加速批准Iovance Biotherapeutics开发的Amtagvi上市,用于治疗晚期黑色素瘤。Amtagvi成为全球首款获批上市的TIL细胞疗法。
与CAR-T类似,TIL细胞疗法属于过继性免疫治疗的一种。TIL疗法从肿瘤中分离浸润的淋巴细胞,体外扩增后回输给患者,由于存在大量患者抗原特异性的TIL细胞,可以特异性识别患者的癌细胞进行杀伤。
Amtagvi定价51.5万美元,2024年第二季度产品总收入为1280万美元,是Amtagvi在美国销售的第一季度。
3
TCR-T疗法
• 全球首款TCR-T细胞疗法——Adaptimmune Therapeutic
2024年8月,Adaptimmune Therapeutics宣布,FDA加速批准其TCR-T治疗产品Tecelra用于治疗无法切除或转移性滑膜肉瘤的成年患者,这些患者既往接受过化疗,HLA-A*02:01、-A*02:02、-A*02:03 或 -A*02:06 呈阳性,且肿瘤表达 MAGE-A4 抗原。
Tecelra是一种针对黑色素瘤相关抗原A4(MAGE-A4)的工程化TCR-T疗法。Tecelra使用每个患者自身的免疫细胞在一次性输注治疗中识别和攻击癌细胞,这与目前晚期滑膜肉瘤的治疗标准有显著不同。
值得一提的是,Tecelra不仅是全球首个获批上市的实体瘤癌症T细胞疗法,也是全球首款获批上市的TCR-T疗法。Adaptimmune计划今年至少开设6到10个授权治疗中心,并在头两年内开设大约30个治疗中心。
4
胰岛细胞疗法
• 首款同种异体胰岛细胞疗法——CellTrans
2024年6月,FDA批准CellTrans开发的同种异体胰岛细胞疗法Lantidra上市,用于治疗1型糖尿病。
Lantidra是FDA批准的首款从逝去供体获得的胰腺细胞生成的同种异体胰岛细胞疗法,用于治疗即使接受强力糖尿病治疗和教育,仍然由于严重低血糖的反复发作,无法达到目标糖化血红蛋白水平的1型糖尿病成人患者。
Lantidra利用死亡捐献者的正常胰岛细胞,通过导管注入到患者的肝门静脉中,从而补充患者体内缺乏的胰岛素和其他激素,增强血糖控制和减少低血糖发作。
5
基因疗法
• 首个获批用于治疗早期CALD的一次性基因疗法——Bluebird
2022年9月,Bluebird宣布,FDA已加速批准Skysona用于减缓患有早期活动性脑肾上腺脑白质营养不良(CALD)的4-17岁男孩神经功能障碍的进展。
Skysona是首个获批用于治疗早期CALD的一次性基因疗法,旨在将ABCD1 cDNA的功能性拷贝添加到患者自身的造血干细胞中,从而产生功能性肾上腺脑白质营养不良蛋白。
财报显示,2023年,Skysona销售额为1240万美元。
• 全球首款可重复给药的基因疗法——Krystal Biotech
2023年5月,FDA批准Krystal Biotech开发的外用凝胶药物Vyjuvek上市,用于治疗6个月或以上的营养不良性大疱性表皮松解症(DEB)患者。
Vyjuvek是首款可重复给药的基因疗法,每周一次局部应用于伤口,也是FDA批准的首个且唯一一个用于治疗隐性和显性DEB的药物。DEB属于罕见的遗传性皮肤病,主要表现为皮肤或黏膜脆性增加,即受到轻微外伤或摩擦后出现破损、水疱或大疱改变。
Vyjuvek的定价约为2.4万美元/瓶,平均每位患者每年使用26瓶。这意味着每位患者每年的费用约为63万美元,经过政府强制折扣后为48.5万美元。2024上半年,Vyjuvek营收1.156亿美元。
• 全球首款DMD基因疗法——Sarepta Therapeutics
2023年6月,Sarepta Therapeutics开发的Elevidys作为全球首款DMD一次性基因疗法获得FDA批准上市,用于治疗4-5岁可独立行走的DMD儿童。
今年6月,FDA批准Elevidy扩展适应症,允许用于治疗4岁及以上必须携带确认的DMD基因突变的DMD患者。其中,对所有4岁及以上确诊DMD基因突变的患者,FDA给予传统批准;对无法行走的患者,给予加速批准。Elevidys禁用于DMD基因第8和/或第9外显子有缺失的患者。
随着辉瑞的同类产品PF-06939926临床开发失败,目前还未有具有潜在竞争力的候选产品。财报显示,Elevidys在2023年销售额超2亿美元,远超预期。2024上半年,Elevidys销售额为2.56亿美元。
• 全球首款血友病A基因疗法——BioMarin Pharmaceutical
2023年6月,FDA批准BioMarin Pharmaceutical开发的Roctavian用于治疗严重A型血友病成人患者。具体适应症为:无因子VIII抑制剂史且未检测到AAV5抗体的重度A型血友病成人患者。
由此,Roctavian成为A型血友病领域的首款基因疗法。Roctavian使用AAV5病毒载体递送表达凝血因子VIII的转基因。它的优势在于,患者可能只需要接受一次治疗,肝细胞就可以持续表达凝血因子VIII,从而不再需要长期接受预防性凝血因子注射。
2024上半年,Roctavian营收0.083亿美元。
• 全球首款CRISPR基因疗法——Vertex&CRISPR Therapeutics
2023年11月,Casgevy获MHRA有条件上市许可,用于治疗12岁及以上镰状细胞病(SCD)与输血依赖性β地中海贫血(TDT)患者,成为全世界首款获批上市的CRISPR基因编辑疗法。
次月,FDA也批准Casgevy用于治疗SCD,定价220万美元。今年1月,该疗法再获FDA批准用于治疗12岁及以上TDT患者。
Casgevy利用CRISPR/Cas9基因编辑系统,在体外对来自患者的造血干细胞进行编辑,使血红细胞生产高水平的胎儿血红蛋白。2015年,Vertex和CRISPR Therapeutics达成战略研究合作,最终推出Casgevy。
• 全球首款MLD基因疗法——Orchard Therapeutics
2024年3月,FDA批准由Orchard Therapeutics开发的Lenmeldy,用于治疗满足特定条件的异染性脑白质营养不良(MLD)儿童患者。此次获批,标志着Lenmeldy成为首个针对MLD的基因疗法。
Lenmeldy旨在通过将一个或多个功能性人类ARSA基因的副本在体外插入患者自身造血干细胞的基因组中,使用慢病毒载体来纠正MLD的潜在遗传原因。修复的基因细胞被输回患者体内,一旦植入,它们会分化成多种细胞类型,其中一些会越过血脑屏障进入中枢神经系统并表达功能性酶。这种方法有望通过一次治疗恢复酶功能以停止或减缓疾病进展。
Lenmeldy在美国的定价为425万美元,刷新“天价药”记录。Lenmeldy上市前,协和麒麟已完成对Orchard的收购。
6
小核酸
• 治疗遗传性ALS的首款疗法——Ionis Pharmaceuticals
2023年4月,FDA加速批准Qalsody(tofersen)用于治疗具有SOD1突变的ALS成人患者,这一适应症是根据Qalsody治疗的患者血浆神经丝轻链的减少而加速批准的。
Qalsody是一种ASO药物,它可以与编码SOD1的mRNA结合,造成其被核糖核酸酶RNase-H降解,进而减少突变的SOD1蛋白生成。这是FDA批准治疗遗传性ALS的首款疗法,也是首款基于生物标志物加速批准的ALS疗法。
2024上半年,Qalsody全球销售额为0.1亿美元。
• 全球首款可通过自动注射器自行给药用于治疗ATTRv-PN的药物——阿斯利康&Ionis Pharmaceuticals
2023年12月,阿斯利康和Ionis联合研发的Wainua(eplontersen)获FDA的批准,用于治疗成人遗传性转甲状腺素蛋白介导的淀粉样变性的多发性神经病。Wainua是首个可通过自动注射器自行给药用于治疗ATTRv-PN的获批药物。
作为一种ASO-GalNAc缀合物,Wainua通过与TTR mRNA结合引起突变型和野生型TTR mRNA降解,从而减少血清TTR蛋白和组织中TTR蛋白沉积。其旨在通过抑制错误折叠突变TTR蛋白的产生来减缓疾病的进展并改善患者的生活质量。患者可自己皮下注射,每月使用一次。
2024上半年,Wainua销售额为0.21亿美元。
2
率先取得监管里程碑的本土创新
已上市产品
• 中国首个获批的CAR-T疗法——复星凯特
2021年6月,复星凯特阿基仑赛注射液已正式获得NMPA批准,这意味着中国迎来首款获批上市的CAR-T疗法。
阿基仑赛注射液是复星凯特从吉利德子公司Kite制药引进的靶向CD19自体CAR-T细胞治疗产品Yescarta,拥有其在中国包括香港、澳门的商业化权利,并于中国境内(不包括港澳台)进行本地化生产。
在完成治疗成人复发难治性大B细胞淋巴瘤的中国境内桥接临床试验后,2020年2月复星凯特在国内提交阿基仑赛注射液上市申请,用于治疗成人复发难治性大B细胞淋巴瘤,包括DLBCL非特指型、原发性纵隔B细胞淋巴瘤(PMBCL)、高级别B细胞淋巴瘤和滤泡淋巴瘤转化的DLBCL,并于3月13日被CDE纳入优先审评。
• 全球首款全人源BCMA CAR-T——驯鹿生物
2023年6月,驯鹿生物申报伊基奥仑赛注射液(商品名:福可苏)获得NMPA批准上市,用于治疗既往经过至少3线治疗后进展(至少使用过一种蛋白酶体抑制剂及免疫调节剂)的复发或难治性多发性骨髓瘤成人患者。
该产品是全球首个获批的全人源BCMA CAR-T细胞治疗产品。
近日,驯鹿生物宣布,伊基奥仑赛注射液用于治疗非肾脏SLE、SLE性肾炎的IND申请已获得FDA的默示许可,这是该产品在自身免疫性疾病领域于中美两国获得的第五个临床批件。
• 中国首款治疗白血病的CAR-T——合源生物
2023年11月,NMPA通过优先审评审批程序附条件批准合源生物的纳基奥仑赛注射液(商品名:源瑞达)上市,用于治疗成人复发或难治性B细胞急性淋巴细胞白血病。这是中国首款白血病治疗领域CAR-T产品,打破白血病30年治疗困境。
申报上市
• 我国首款申报上市的干细胞新药——铂生卓越生物
2024年6月,铂生卓越生物“艾米迈托赛注射液”NDA正式获NMPA受理并被纳入优先审评品种。据悉,这是我国首款申报上市的干细胞新药,其申请的临床适应症为激素失败的急性移植物抗宿主病(aGVHD)。
aGVHD是一种罕见的危及生命的疾病,发生在同种异体造血干细胞移植(HSCT)后,由于供体的免疫细胞攻击宿主的健康细胞导致。该病通常影响皮肤、胃肠道和肝脏。胃肠道急性移植物抗宿主病是HSCT后发生死亡的主要原因。
值得一提的是,在2004年,赵春华教授所在的协和基础所注册申报的中国也是全球第一个干细胞新药正式获准进入临床试验。2006年,其团队研发的第二个干细胞新药“间充质干细胞心梗注射液”也获得了临床批件。两个新药临床批件适应症涵盖了以移植物抗宿主病(GVHD)和心脑血管疾病为代表的难治性疾病。“骨髓原始间充质干细胞”于2006年4月获得II期临床批件,在2020年获批进入III期临床,是国内第一个研发进展进入III期阶段的干细胞药物。
• 国内首个递交NDA的AAV基因治疗药物——信念医药
2024年7月,信念医药宣布,NMPA已正式受理BBM-H901注射液用于治疗血友病B成年患者的新药上市申请。
BBM-H901注射液以静脉给药的方式将人凝血因子IX基因导入血友病B患者体内持续表达,提高并长期维持患者的凝血因子水平,用于预防血友病B成年患者出血。
2022年,BBM-H901注射液被CDE纳入“突破性治疗品种”名单。2024年,该款产品成为国内首个递交新药上市申请的AAV基因治疗药物。
获批IND
1
TIL疗法
• 国内首个进入临床的TIL疗法——沙砾生物
2023年4月,由沙砾生物开发的TIL疗法GT101注射液正式获得NMPA的临床试验默示许可,成为国内目前第一个也是唯一一个进入关键临床阶段的TIL产品。
GT101注射液通过从患者体内获取肿瘤组织并提取TIL,再经过沙砾生物StemTexp®生产平台扩增至10亿量级后输注回患者体内以对抗肿瘤。
• 全球首个采用无需清淋级预处理、无需IL-2注射的TIL疗法——君赛生物
砂砾生物之后,君赛生物的GC101 TIL疗法仅隔2天获得IND批准,全球首个采用无需清淋级预处理、无需IL-2注射临床方案,在多种类型实体瘤的I期临床试验中展现良好疗效。据悉,GC101 TIL疗法首个优选适应症即将进入关键II期临床。
• 全球首款进入临床的肝癌TIL产品——百吉生物
BST02是百吉生物全球首款针对肝癌的TIL产品,自2024年1月同时获得中美临床批件,7月完成中美双报双批临床I期首例受试者入组。
作为基于患者自身肿瘤浸润淋巴细胞扩增的T细胞治疗产品,BST02属于过继性免疫细胞治疗技术,具有治疗所有类型肝癌的潜力。与传统的TIL药物相比,BST02具有可冻存、突破距离限制、无需高剂量白介素-2辅助用药等优势。
• 全球首款非病毒载体基因修饰TIL疗法获批IND——君赛生物
GC203是君赛生物自主研发的全球首款非病毒载体基因修饰TIL疗法,是天然TIL细胞的“加强版”,使TIL细胞稳定产生可自聚集的膜结合细胞因子IL-7,其临床试验已于2024年4月获国家药监局批准。
GC203以天然TIL细胞为基础,通过非病毒载体介导的基因工程技术,使TIL细胞稳定表达膜结合型、可自发聚集的细胞因子。生产过程无需滋养细胞,工艺更简便;基因修饰过程,采用非病毒载体,成本更低,安全性更高,修饰效率与病毒载体相当(平均45%)。
2
CAR-T疗法
• 全球首款利用基因沉默技术而降低CRS和ICANS的CAR-T进入临床——优卡迪生物
U01(CD9-CART-shRNA-IL6)是全球唯一通过小干扰RNA基因沉默技术而降低CRS和ICANS的新一代CAR-T产品,已于2020年获得CDE批准进入临床。
U01除能治疗除难治复发B-ALL外,还可以被批准容许治疗“脑白”患者,目前正处于临床Ⅱ期。据悉,这是国际上首款打破“脑白”这一禁忌症的CAR-T疗法,对白血病中枢浸润的治疗具有重要意义。
• 全球首个获批进入临床的T细胞系恶性肿瘤自体CD7-CAR-T疗法——博生吉医药
2021年8月,由博生吉医药自主研发的自体CD7-CAR-T产品(PA3-17注射液)获CDE的IND批准,用于治疗难治/复发T淋巴母细胞白血病/淋巴瘤。这是全球首个以T细胞系肿瘤为适应症被批准进入临床的自体CD7-CAR-T产品,2021年11月和2022年12月分别被FDA和欧盟委员会(EC)授予了孤儿药资格认定。
PA3-17注射液是一款基于CD7纳米抗体开发的自体CAR-T产品,采用非基因编辑策略攻克了诸多CMC挑战,并开发成功了高度优化的全自动制备工艺。目前注册临床一期剂量爬坡试验已完成。
• 首款非病毒快速制备的CAR-T疗法获批IND——星尘生物
2023年10月,星尘生物宣布其非病毒快速制备的FIT-CD19 CAR-T免疫疗法治疗非霍奇金淋巴瘤已获FDA对IND申请的批准。
这一创新性疗法的独到之处在于采用非病毒制备技术,将制备时间缩短至仅需两天,而成本仅为目前上市产品的几分之一。这一突破性技术的成功应用,不仅提高生产效率,降低治疗成本,也为更广泛的患者提供一种更为便利、经济的治疗选择。
• 首个中美双报获批IND靶向EpCAM的CAR-T产品——易慕峰
2024年2月,易慕峰自体CAR-T细胞注射液产品(IMC001)IND申请临床试验申请在中国CDE批准之后,又获得了美国FDA的临床研究许可,用于治疗上皮细胞黏附分子(EpCAM)表达阳性的晚期消化系统肿瘤,包括但不限于晚期胃癌(GC)/食管胃结合部腺癌(GEJ)。EpCAM CAR-T可以同时靶向原发灶、CTC、转移灶。
IMC001成为全球首个中美双报获批IND的靶向EpCAM的CAR-T产品,已在IIT研究中展示出良好的安全性和有效性。
• 全球首创中美双报双批靶向EB病毒抗原CAR-T——百吉生物
2024年8月,百吉生物自主研发的BRG01注射液(EBV特异性CAR-T)获得FDA批准其开展关键性II期临床试验。
数据显示,BRG01展现出了卓越的安全性和初步有效性:75%高剂量患者肿瘤病灶缩小且代谢活性降低。部分患者甚至实现了肿瘤病灶的100%完全缓解。此外,BRG01还表现出卓越的抗EB病毒效力,患者回输后外周血中的EB病毒载量显著降低至正常水平。
• 全球同类首创CLDN18.2 CAR-T完成II期入组——科济药业
2024年8月,科济药业宣布,全球同类首创、靶向CLDN18.2自体CAR-T细胞候选产品舒瑞基奥仑赛注射液,在中国进行的针对晚期胃癌/食管胃结合部腺癌的确证性II期临床试验,已按照临床方案完成全部受试者的入组和随机。
2022年3月,CT041于北京大学肿瘤医院完成了中国确证性Ⅱ期临床试验的首例患者入组,主要治疗既往接受过至少二线治疗失败的CLDN18.2表达阳性的晚期胃癌/食管胃结合部腺癌。
CT041成为全球首个进入到确证性Ⅱ期临床试验的用于治疗实体瘤的CAR-T细胞候选产品;也是全球首个已获得FDA和CDE以及加拿大卫生部的IND/CTA批准、并正在进行临床试验研究的靶向CLDN18.2的CAR-T细胞候选产品。
3
CAR-NK疗法
• 国内首个获FDA IND批准的非基因修饰异体外周血NK疗法——恩凯赛药
2024年1月,由恩凯赛药开发的NK010获得FDA I期临床试验许可,首选卵巢癌适应症进行探索。该产品为国内首个获得FDA临床批准的非基因修饰异体外周血NK细胞药物。
NK010具有受体谱最优化、靶点多样化、高纯度、普适性等高抗癌优势,使其具有治疗多类型肿瘤的潜力。NK010也存在扩展至非肿瘤疾病治疗的潜在空间,还是后续一系列合成NK细胞药物的最佳底盘细胞。
• 国内首个通用型CAR-NK治疗SLE获批IND——先博生物
2024年2月,先博生物自主研发的靶向CD19的嵌合抗原受体基因修饰的NK细胞注射液正式获得CDE的临床默示许可,适应症为中重度难治性系统性红斑狼疮(SLE)。这是SLE适应症在国内首个通用型NK产品获批IND。
通用型CAR-NK疗法,与其它创新细胞治疗产品相比显著降低患者的经济负担并且即时可用。叠加NK细胞突出的安全性优势,让NK疗法有望在不断发展的自身免疫性疾病领域脱颖而出。
• 国内首个获批临床的通用型实体瘤细胞疗法——羿尊生物
2024年7月,由羿尊生物自主开发的CNK-UT002细胞注射液的IND成功获得CDE默示许可,适应症为晚期实体瘤 (包括但不限于肝癌和黑色素瘤),这是国内首个获批临床的通用型实体瘤细胞疗法
CNK-UT002细胞注射液采用的是一种集成了CNK-T和U-T两大平台技术的通用型细胞技术,采集健康供者的T细胞经基因编辑改造后,赋予NK细胞识别功能并具有T细胞的激活打击能力,能特异性地识别和治疗各种实体和血液肿瘤,同一款CNK-UT细胞药物可实现对肝癌、黑色素瘤、结直肠癌、肾癌、T细胞白血病等多种实体肿瘤和白血病细胞的杀伤。
• 国内首款获批IND的非基因修饰同种异体NK疗法——希灵生物
2024年7月,希灵生物的“CEL001注射液”获得临床试验默示许可,适应症为晚期实体肿瘤。这是国内首款批准IND的非基因修饰同种异体NK细胞候选产品。
CEL001注射液来源于健康人外周血,是基于体外培养技术,经过分离、活化、增殖等步骤,获得的一类具有高表达激活性受体、高水平表达颗粒酶、穿孔素、兼具细胞因子分泌功能和细胞杀伤作用的通用现货型NK细胞候选产品。
• 3款首创同源异体现货通用型非病毒载体非基因编辑的抗体NK偶联药物进入临床——英百瑞
1. IBR854是全球首款进入注册临床阶段靶向5T4治疗实体瘤的抗体偶联NK细胞产品(2023年1月获得CDE临床默示许可)。
2. IBR733是全球首款双靶向CD33/CLL1治疗AML进入注册临床阶段的抗体偶联NK细胞产品(2023年12月获得CDE临床默示许可)。
3. IBR822是全球首款进入注册临床阶段靶向Trop2治疗实体瘤的抗体偶联NK细胞产品(2024年4月获得CDE临床默示许可)。
4
iPS疗法
• 全球首款进入注册临床的iPSC衍生通用型帕金森细胞治疗产品——睿健医药
2023年8月,由睿健医药通用型细胞治疗产品NouvNeu001(人源多巴胺能前体细胞注射液)获得NMPA批准进入I/II期联合临床,用于治疗中晚期帕金森病。这是全球首款进入注册临床的iPSC衍生通用型帕金森细胞治疗产品,也是全球首个进入临床阶段的基于化学诱导的通用型细胞治疗产品。
2024年1月,NouvNeu001在北京医院完成了注册临床阶段首例患者给药。目前已经进入剂量拓展阶段。2024年6月,NouvNeu001获得FDA批准进入海外临床I期。同时,睿健开创性产品被FDA授予特别豁免权,也是全球唯一获得FDA“特殊豁免”的通用型细胞治疗产品。
• 中国首例临床级iPS衍生细胞移植治疗帕金森病给药——士泽生物
2024年1月,士泽生物临床级自体iPSC衍生多巴胺能神经前体细胞注射液经颅内立体定位注射微创手术方式移植治疗帕金森病,在上海市东方医院(同济大学附属东方医院)成功完成。据悉,这是中国首例临床级iPS衍生细胞移植治疗帕金森病,也是中国首例、世界第二例自体iPS衍生细胞替代性移植治疗帕金森病。
该项目是我国首个正式获批的iPS衍生细胞治疗神经系统疾病的国家级干细胞备案临床研究项目。手术在两小时以内完成:首例受试者接受临床级iPS衍生细胞移植后,无手术及围术期的并发症或其他不良安全事件,各项检测指标正常且已顺利出院,平稳度过观察期并进入正式随访期。
• 全球首个ipsc衍生前脑神经前体注射液获中美IND批准——霍德生物
2024年3月,霍德生物开发的iPSC衍生异体通用型前脑神经前体细胞注射液hNPC01,在30天默许期内提前收到FDA通知可以开展针对缺血性脑卒中偏瘫后遗症的I/IIa注册临床试验,无附加条件。
hNPC01已知是目前全球首个进入注册临床的多能干细胞(包括iPSC及胚胎干细胞ESC)衍生的前脑神经前体细胞产品,同时也是中国首个原研并成功获得美国IND批准的多能干细胞衍生产品(包含衍生间充质样、神经、心肌、免疫细胞、胰岛细胞等所有品类)。
5
TCR-T疗法
• 国内首个获批IND的TCR-T疗法——香雪制药
2024年7月,CDE同意香雪制药的TAEST16001注射液纳入突破性治疗品种名单,标志着该产品作为中国第一个IND获批开展临床试验的TCR-T细胞治疗新药取得重大突破。
TAEST16001注射液是香雪制药的一款靶向NY-ESO-1的亲和力增强的TCR-T细胞免疫治疗产品,其首个临床研究适应症是HLA-A*02:01阳性且表达NY-ESO-1抗原的晚期软组织肉瘤患者。该产品是中国首个获得IND批件的TCR-T产品,也是中国唯一进入II期临床的TCR-T产品。
• 全球首款实体瘤的TCR-T产品——百吉生物
BRL03是百吉生物自主研发的全球首款针对肺癌和胃癌等多种实体肿瘤的TCR-T产品,I期临床试验也在今年6月完成首例受试者入组,并计划在今年年底完成所有I期受试者入组。
BRL03是第四代TCR-T细胞疗法,是基于其自主创新技术平台IDENTIFIER®开发的,能够针对多种实体瘤。而另一个自主研发的平台MSE-T使得BRL03具有额外的功能模块组件,减少细胞耗竭以提升抗肿瘤的持久性。
6
基因疗法
• 首个戈谢病AAV基因疗法获批IND——凌意生物
2024年1月,凌意生物旗下LY-M001注射液的IND申请获得NMPA的默示许可。这是中国首个自主研发,针对I型或III型戈谢病的AAV基因治疗药物。该产品使用重组腺相关病毒rAAV为载体,通过单次静脉输注给药后即可表达患者所需的葡萄糖脑苷脂酶。去年10月,该产品获得美国FDA孤儿药资格认定,用于治疗戈谢病。
• 首个采用“两质粒包装系统”的AAV基因疗法获批IND——金唯科生物
2024年2月,金唯科生物研发的针对新生血管性年龄相关性黄斑变性(nAMD)基因治疗产品“JWK001注射液”的IND申请获得NMPA批准。
J这是首个采用“两质粒包装系统”的AAV基因疗法,通过视网膜下给药将承载着全新自主设计的抗VEGF蛋白表达框的AAV递送至眼部细胞中,可持续高效表达抗VEGF蛋白。JWK001注射液设计多靶点基因表达框,增加覆盖单一靶点不敏感患者,提升药物疗效。
• 国内首个LNP-mRNA体内基因编辑药物获批临床——尧唐生物
2024年3月,尧唐生物宣布,CDE批准了YOLT-201的临床试验申请,这也是国内首个采用脂质纳米颗粒(LNP)介导的体内基因编辑药物获得临床试验批准。
YOLT-201通过LNP以mRNA形式递送CRISPR-Cas9基因编辑系统,在人肝脏中实现高效特异的转甲状腺素蛋白(TTR)靶基因编辑,以永久性降低血液中的TTR蛋白水平,有望实现转甲状腺素蛋白淀粉样变(ATTR)的终身治疗,包括转甲状腺素蛋白淀粉样变性多发性神经病(ATTR-PN)和转甲状腺素蛋白心脏淀粉样变(ATTR-CM)。
• 国内首个获批临床的碱基编辑疗法——正序生物
2024年4月,正序生物CS-101注射液获得NMPA的IND批准,正式启动了临床I期试验,成为国内首个获批临床的碱基编辑疗法。同月,正序生物入组了首位临床试验患者。
CS-101的原理是通过采集患者自体造血干细胞,利用高精准变形式碱基编辑器tBE(transformer Base Editor),对患者自体造血干细胞中的HBG1/2启动子区域进行精准碱基编辑,模拟健康人群中天然存在的有益碱基突变,重新激活γ-珠蛋白的表达,重建血红蛋白的携氧功能。
7
溶瘤病毒
• 国内自研首个获批临床的溶瘤痘苗病毒产品——华药康明
2022年9月,华药康明的溶瘤痘苗病毒产品KM1的IND申请获得NMPA批准。2022年12月,KM1的IND申请获得FDA批准。
这是国内自研首个获得临床批件的溶瘤痘苗病毒产品。该产品携带免疫调节基因IL-21和4-1BBL,旨在更好地激活抗肿瘤的免疫反应。目前KM1在中国开展I期临床试验中。
• 全球首款静脉疱疹溶瘤产品完成美国临床I期——亦诺微医药
2023年11月,亦诺微医药宣布,全球首款处于临床阶段的静脉疱疹溶瘤病毒产品MVR-T3011 IV,已在美国成功完成针对多个晚期大瘤种适应症进行的临床I期试验。
MVR-T3011是亦诺微医药拥有自主知识产权的三合一疱疹溶瘤病毒创新产品,其基于对野生HSV-I型疱疹病毒骨架的全新设计,确保病毒在肿瘤细胞内拥有强劲的复制能力的同时,在正常细胞内复制能力得到抑制,实现最优减毒效果。此外,MVR-T3011携带了两个最新且被充分验证过的外源性免疫调节基因——PD-1抗体和IL-12,以促进肿瘤微环境的免疫反应。
IND获受理
1
基因疗法
• 全球首个用于GA-I的基因治疗产品IND获受理——天泽云泰
2023年4月,据CDE官网公示,天泽云泰的基因治疗产品“VGM-R02b”新药临床试验申请获得受理。
据悉,VGM-R02b是一款用于治疗戊二酸血症I型(GA-I)的基因治疗产品,属于儿童专用创新药。2022年5月,该产品已被FDA授予用于治疗GA-I的罕见儿科疾病认定(RPDD)资格。同时,VGM-R02b也是全球首个用于GA-I的基因治疗产品,将有机会以罕见儿科疾病产品申请美国儿科孤儿药优先审评券(PRV)。
2
CAR-T疗法
• 全球首个针对CLDN18.2表达阳性的晚期实体瘤恶性腹腔积液治疗的细胞药物产品IND获受理——华道生物
2024年8月,华道生物生产的HD004CAR-T细胞于获CDE正式受理。
HD004CART是全球首个针对CLDN18.2表达阳性的晚期实体瘤恶性腹腔积液治疗的首个细胞药物产品。
识别微信二维码,添加生物制品圈小编,符合条件者即可加入
生物制品微信群!
请注明:姓名+研究方向!
版
权
声
明
本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(cbplib@163.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。
细胞疗法免疫疗法基因疗法财报上市批准
2024-08-14
·药通社
论坛导览-点击查看大图↓↓
*议程更新中,以现场为准
B馆-生物馆
GLP-1及多肽论坛
B馆-B102 8月15日
▼下滑浏览全部
主持人:李湘,中肽生化创始人&董事长
▷09:00-09:25
多肽类药物在临床期间的CMC研究的多变性
苑字飞甘李药业,副总经理,首席技术官
▷09:25-09:50
GLP-RAs新药研发趋势-长效化、多靶点
陈小新众生睿创,CEO
▷09:50-10:15
创新型靶向GLP-1R/GIPR的双靶点激动剂的发现和临床转化
甘雨龙道尔生物,临床运营高级总监
▷10:15-10:40
iCVETide多肽新药发现平台技术及实用案例分享
孟广鹏成都诺和晟泰,创新药总监
▷10:40-11:05
多肽药物的质量控制策略
付玉清 健元医药,高级研发总监
▷11:05-11:30
GLP类药物研发进展及商业化前景
章 华鸿运华宁,研发高级总监
▷11:30-11:55
GLP-1药物的差异化发展策略
谢 天甘李药业,医学事业部负责人
▷11:55-12:20
圆桌对话:多肽治疗领域扩展及出海机会
李 湘 中肽生化创始人&董事长(主持人)
陈小新众生睿创,CEO
孙汉栋九源基因,副总经理徐洪岩吉尔多肽,董事长
▷12:20-13:30
午餐
主持人:赖锦昌,美迪高,总经理
▷13:30-13:55
GLP-1,除了减重,还能卷什么?
左亚军 仁会生物,总经理
▷13:55-14:20
司美格鲁肽与替尔泊肽杂质鉴定研究案例分享
刘国柱 长沙晨辰医药,创始人&董事长
▷14:20-14:45
新一代人源超长效GLP-1药物创新研发之路及关键临床数据解读
徐文洁银诺医药,高级副总裁&首席商务官
▷14:45-15:10
多肽实体文库高通量筛选体系及特色应用开发
林 定禾泰健宇生物,研发总监
▷15:10-15:35
伊匹乌肽-替代糖皮质甾体激素的广谱抗炎原创药
夏献民益承生物,董事长&总经理
▷15:35-16:00
EMPHASES多肽合成技术及在药物多肽合成中的应用
郑庆泉 同隽医药,创始人&董事长
▷16:00-16:25
环肽创新药的发现:从长效棘白菌素到口服PCSK9抑制剂
原晨光祥根生物,联合创始人&CSO
立即扫码报名
XDC领域的差异化创新和国际化策略
B馆-B103 8月15日
▼下滑浏览全部
大会主席&主持人:夏明德,英诺湖医药,CEO&董事长
▷08:55-09:00
大会主席致辞
夏明德英诺湖医药,CEO&董事长
▷09:00-09:20
ADC FIC 药物因CMC的生产缺陷导致批准上市延缓的启示
刘翠华百奥泰生物,高级副总裁
▷09:20-09:40
小型多肽类及双靶向偶联药物的研究、临床开发进展和成药展望
邵 军同宜医药,首席技术官
▷09:40-10:00
宜联生物TMALIN ADC技术平台:提高药效、在新空间寻找靶
蔡家强宜联生物,联合创始人&CSO
▷10:00-10:20
MIDD视角下的ADC药物开发
苏 霞康龙化成,临床药理&定量药理高级总监
▷10:20-10:40
未来已来:端到端的创新ADC药物发现与商业化开发模式
秦 刚启德医药,CEO
▷10:40-11:00
核药临床试验设计的特殊性与实施的挑战
刘 艳米度生物,首席医学官
▷11:00-11:20
ADC的组合创新
谢雨礼 上海微境生物,创始人&总经理
▷11:20-12:00
圆桌:如何从早期设计降低XDC的临床失败?
1.XDC从早期设计到临床开发成败案例
2.失败的原因分析
3.从失败的案例中可以得到哪些启发?如何避免
夏明德英诺湖医药,CEO&董事长(主持人)
刘翠华百奥泰生物高级副总裁
邵 军同宜医药,COO
蔡家强宜联生物,CSO
秦 刚启德医药,CEO肖志华奥浦迈,董事长
▷12:00-13:30
午餐
主持人:刘艳,米度生物,首席医学官
▷13:30-13:50
生命科学前沿应用之ADC药物研发
徐雍羽 瑞孚迪,类器官和生物制药项目经理
▷13:50-14:10
New Drug Modality在未被满足临床上的探索--放射性偶联药物
单 波博锐创合 ,CEO
▷14:10-14:30
创新核药研发与产业链之诺宇解决方案
方 鹏诺宇医药,董事长&创始人
▷14:30-14:50
靶向Nectin4核素偶联药物的开发
习 宁范恩柯尔,创始人&CEO
▷14:50-15:10
核药产业发展现状
邓启民成都云克药业,总经理
▷15:10-15:30
靶向GPC3放射性药物的筛选与开发
宋 征艾博兹医药,临床前开发副总裁
▷15:30-15:50
核素药物全球化专利布局挑战与策略
唐华东植德律所,合伙人
立即扫码报名
抗体掌门人对话联合主办:壹生科 学术支持:上海市生物信息学会智能药物专委会
B馆-B101 8月15-16日
▼下滑浏览全部
8月15日上午主持人:陈明久,博奥信创始人、董事长兼首席执行官
▷08:55-09:00
联合主办方致辞马宁宁 沈阳药科大学,教授;壹生科,董事长&创始人
▷09:00-09:30
可变区定点偶联技术(OC-BODY)在单抗和双抗中的应用
马宁宁 沈阳药科大学,教授;壹生科,董事长&创始人
▷09:30-10:00
基于AI的抗体发现与设计
曹志伟复旦大学,教授
▷10:00-10:30
ADC早期研发的生物学和DMPK评价李黎爱思益普生物,VP
▷10:30-11:00
复宏汉霖的全球化战略
盛严慈复宏汉霖,生物制药全球战略与项目管理总经理
▷11:00-12:00
圆桌:如何走出差异化生物科技企业出海之路?
主持人:马宁宁沈阳药科大学,教授;壹生科,董事长&创始人
唐阳刚 丽珠集团,总裁
高长寿 艾力斯医药,首席技术官
陈立模 女王之舟药业,董事长&CEO
滕毓敏 赛洱纳博生物,联合创始人&CEO
华 烨 烨辉医药,董事长兼首席执行官
▷12:00-13:30
午餐
8月15日下午主持人:刘江海,成都盛世君联,CEO
▷13:30-14:00
抗病毒单克隆抗体药物研发进展
严景华昌平实验室,新型疫苗和抗体药物领域领衔科学家
▷14:00-14:30
HER2抗体偶联药物诱导间质性肺炎的生物学机制研究
郭鹏中科院杭州医学研究所,教授
▷14:30-15:00
抗体免疫学基础研究进展
孟飞龙中科院分子细胞科学卓越创新中心,研究员
▷15:00-15:30
疼痛领域抗体药物机会
王春河达石药业,董事长 &创始人
▷15:30-16:00
纳米抗体早研的案例分享和思考
刘江海成都盛世君联,CEO
8月16日主持人:曹志伟,复旦大学,教授
▷09:00-09:20
研究者视角看ADC药物研发现状及问题
薛俊丽上海东方医院,教授
▷09:20-09:40
ADC药物应用场景和抗体的选择
李元浩荣昌生物,副总经理
▷09:40-10:00
双特异性纳米抗体介导的胞内蛋白靶向降解
马兴元华东理工大学 教授、博导
▷10:00-10:20
创新的抗体-细胞因子融合蛋白的研发
周海平徕科生物,CEO
▷10:20-10:40
高覆盖度人源膜蛋白兔单克隆抗体的制备及应用
王平致知生物,首席商务官
▷10:40-11:00
抗体偶联药物应用的临床进展
韦青浙江省肿瘤医院,胃胰肠肿瘤内科副主任医师
▷11:30-12:00
圆桌:多种类型抗体相继出现,未来抗体的演进方向在哪里?
主持人:马宁宁 沈阳药科大学,教授;壹生科,董事长&创始人
李新芳 迈百瑞,CEO
陈明久 博奥信创始人、董事长兼首席执行官
翁志兵 三生制药,CDMO事业部总经理
杜 新 埃格林医药,联合创始人&CEO
董 欣 逻晟生物,创始人&董事长&CEO段 清 东曜药业,药物研发及技术开发负责人
陈建新 臻格生物,董事长&CEO
立即扫码报名
核酸药物开发前沿论坛
B馆-B102 8月16日
▼下滑浏览全部
主持人:冯冰,中肽生化,寡核苷酸研发资深总监
▷09:00-09:20
mRNA疫苗与mRNA药物
回爱民惠正奇医药,创始人
▷09:20-09:40
靶向mRNA的碱基编辑技术开发与应用进展
忻 蓉时夕生物,总经理
▷09:40-10:00
小核酸药物的临床前药代动力学研究
刘文玥 华西海圻,药代技术总监
▷10:00-10:20
小核酸药物递送技术-偶联递送平台探索
张佩琢吉玛基因,董事长
▷10:20-10:40
寡核苷酸药物中的杂质来源
冯 冰中肽生化,寡核苷酸研发资深总监
▷10:40-11:00
小核酸药物的创新实践
苏晓晔石药集团,核酸药物研究院负责人
▷11:00-11:20
Nucleosidic phosphoramidites: an optimized approach in their synthesis, quality evaluation and impurities characterization
Beatrice Trucchi Flamma S.p.A. - Italy R&D Project Manager
▷11:20-11:40
BH-RACETM新型核酸药物平台
胡 翰滨会生物,副总裁
▷12:00-13:30
午餐
主持人:张力,北京瑞博奥医药科技有限公司,副总经理
▷13:30-13:50
小核酸干扰药物的递送和临床进展
袁旭东艾康药业,CEO
▷13:50-14:10
治疗肝和肺部疾病的siRNA全新分子设计
黄 毅国为医药,研究院副院长
▷14:10-14:30
全和诚核酸药物递送平台(拟)
周经经全和诚,研发副总经理
▷14:30-14:50
激发小核酸长效性潜力开发慢性病案例(拟)
万金桥先衍生物,总经理
▷14:50-15:10
AI驱动的高效mRNA疫苗开发
费才溢澄实生物,vp&联合创始人
▷15:10-15:30
双靶向核酸适体技术
王雪强中国科学院杭州医学研究所,研究员
▷15:30-15:50
寡核苷酸药物的DMPK的研究挑战和案例分享
陈桂英 宏韧医药,化学药物生物分析部门总监
立即扫码报名
创新药早期开发论坛
B馆-B103 8月16日
▼下滑浏览全部
策划人&主持人:张水华,新樾生物,副总经理
▷09:00-09:25
铁死亡研究的突破与新疗法的涌现
徐 峻中山大学,教授
▷09:25-09:50
Smart DEL(DEL及AI相结合的)的筛选及案例分享
金 锋新樾生物,副总经理
▷09:50-10:15
Explore New DEL Modality Beyond Binding Assay
黄已然广东省小分子新药创新中心,新药筛选中心副总监
▷10:15-10:40
勃林格殷格翰在生物医药前沿创新进行外部合作的战略方向
韩奇峰勃林格殷格翰,全球业务拓展及许可部门副总监
▷10:40-11:05
新药发现新技术的趋势和各自优势及特点
张 强浙江大学教授;山东博观首席科学家
▷11:05-11:30
创新药立项评估与药物早期发现过程中的关键点
周厚江海正药业,研发中心副总经理、创新药研究院院长
▷11:30-12:15
圆桌对话:药物早期研发阶段的主要目标是什么?什么样的早期药物研发是成功的?
贺 耘 深圳湾实验室百瑞创新中心主任胡 畏礼来中国,创新合作中心资深总监
边 峰BMS,全球药物研发中国综合科学团队执行总监
方剑武大睿生物,副总裁
吴宏忠分子之心,商务副总裁
▷12:15-13:30
午餐
靶向蛋白降解药物开发
策划人&主持人:曾雳 和径医药首席执行官
▷13:30-13:55
万物皆可降:靶向蛋白降解药物研发思路探讨
冯 焱领泰生物,CEO
▷13:55-14:20
蛋白降解GlueTacs®平台构建及其肿瘤及免疫疾病治疗领域的药物开发
杨小宝标新生物,创始人、董事长兼首席执行官
▷14:20-14:45
Bi-XDC技术及临床研究进展
王贵涛同宜医药副总经理、研究院院长
▷14:45-15:10
分子伴侣介导的蛋白降解技术平台
英伟文珃诺生物创始人兼首席执行官
▷15:10-15:35
基于肿瘤组织特异性E3连接酶的PROTAC药物开发
张继跃奥瑞药业,联合创始人兼首席执行官
▷15:35-16:15
圆桌讨论:蛋白降解技术的技术开发趋势和挑战?
曾 雳和径医药首席执行官(主持人)
冯 焱领泰生物,CEO
杨小宝标新生物创始人、董事长兼首席执行官
英伟文珃诺生物创始人兼首席执行官
胡逸民 Chimergen Tx ,联合创始人兼CSO
立即扫码报名
SynBio Suzhou中国合成生物学“科学家+企业家+投资家”大会
B馆-B104+105 8月15-16日
▼下滑浏览全部
论坛一 农业、食品与能源
▷09:30-10:00
大宗醇酸非粮合成生物制造引领者
杨世辉湖北大学教授,省部共建生物催化与酶工程国家重点实验室副主任,武汉睿嘉康生物科技有限公司创始人
▷10:00-10:30
风口正当时——麦角硫因在各领域应用及科学依据
何儒俊仅三生物创始合伙人兼原料副总
▷10:30-11:00
乳制品企业在生物制造新质生产力领域的投资布局和思考
胡智渊蒙牛创投基金总经理
▷11:00-11:30
合成生物学赋能辅酶系列产品的开发和产业化
张琦邦泰生物创始人兼CSO
▷11:30-12:00
微藻源左旋虾青素的生物合成
屈玉娇元育实验室(Protoga Labs)负责人,深圳市孔雀人才
▷11:30-12:00
食品合成生物学的产业化先驱-HMOs
方诩山东恒鲁生物科技有限公司创始人兼首席科学家
论坛二 医美、医药、大健康
▷13:30-14:00
基于植物合成生物学的化妆品原料创新和应用
王玉亮上海交通大学植物生物技术研究中心副主任、上海交通大学农业与生物学院硕士研究生导师
▷14:00-14:30
益生菌益生元在特殊营养的创新研发和临床实证数据
曾哲达能全球研发中心(荷兰乌特勒支)高级科学家 &亚太区益生菌(科学事务)负责人
▷14:30-15:00
细胞外基质蛋白组合(ECMP)在美妆护肤领域应用及问题皮肤解决方案
赵俊合成生物蛋白安徽省联合共建学科重点实验室主任
▷15:00-15:30
珍稀药用植物天然产物的微生物人工合成
刘晓楠中国科学院天津工业生物技术研究所 副研究员
▷15:30-16:00
负碳生物合成 Synthetic biology for carbon-negative biosynthesis
李恒润光玥生物研发总监
▷16:00-16:30
合成生物学在医美领域创新性突破-从基因定制到美丽再生张骊元一生物创始人
▷16:30-17:30
圆桌讨论:合成生物学面临的监管挑战
论坛三 生物智造
▷09:30-10:00
人工智能蛋白质领域的研究(拟)
洪亮上海交通大学自然科学院与物理天文学院与药学院教授
▷10:00-10:30
人工智能在代糖领域的应用
高航注册金融分析师,复星锐正资本董事总经理
▷10:30-11:00
芯片式高通量DNA合成
董一名芯宿科技联合创始人&CSO
▷11:00-11:30
生成式AI大模型驱动的蛋白质研发与生产
唐建百奥几何创始人兼CEO
▷11:30-12:00
AI技术蛋白质相互作用预测及其应用研究
常珊 江苏理工学院生物信息与医药工程研究所所长
论坛四 技术、工具
▷13:30-14:00
合成生物学产业化平台
郭宏明江苏惠利生物董事长
▷14:00-14:30
AI酶改造与生物制造降本增效
卢星宇天鹜科技CMO
▷14:30-15:00
酶进化技术重塑API中间体合成生态
刘想镁孚泰生物CEO
▷15:00-15:30
酶的改造和固定化技术再中间体和原料药制备中的应用
竺伟尚科生物总经理
▷15:30-16:00
基于脆弱拟杆菌的创新活体生物药开发
李平知易生物副总经理,广东省活体生物药工程技术研究中心副主任
▷16:00-16:30
谷胱甘肽的新应用
王勇 诺云生物创始人&CEO
▷16:00-16:30
Synbio Suzhou合成生物学杰出企业 颁奖仪式
论坛五 优质项目路演
▷09:30-9:50
微生物发酵法高效生产红景天苷
▷9:50-10:00
HMO项目
▷10:00-10:15
植物多酚增效及紫酚生菜食药同源的开发和产业价值
▷10:15-10:30
植物合成生物学--生物医药的绿色可持续生产
▷10:30-10:45
普瑞森合成微生物平台
▷10:45-11:00
合成生物育种——芳香原料项目
▷11:00-11:15
生物法长碳二元酸及长链尼龙产业化项目
▷11:15-11:30
生物基绿色新材料助力可持续发展
▷11:30-11:45
基于纳米机器人智能极限检测技术的标签数据AI决策技术产品
▷11:45-12:00
长效植入剂及医美针剂的产业化
论坛五 商业化与后处理
▷09:30-10:00
生物制造过程控制和优化(拟)方柏山厦门大学教授
▷10:00-10:30
合成生物学使能工具助力酵母细胞工厂创建史硕博北京化工大学教授
▷10:30-11:00
面向生物发酵过程智能监控的分离传感膜及仪器开发
储震宇南京工业大学材料化学工程国家重点实验室教授
▷11:00-11:30
合成生物学产物的分离与纯化张庆武北京日新远望科技发展有限公司总工程师、教授及研究员级高级工程师
▷11:30-12:00
新型分离技术应用
立即扫码报名
国企与Biotech项目合作闭门对接会
会议主持:蔡学钧,前BMS,全球研发副总裁B馆-B106 8月15日
▼下滑浏览全部
▷10:00-10:30
当前形势下,国企和Biotech面临的挑战和机遇
蔡学钧前BMS,全球研发副总裁
▷10:30-10:50
新质生产力时代,国企该如何释放创新潜力和合作机会
▷10:50-12:00
圆桌对话:政策激励下,国企及Biotech各自的机遇和挑战
密集生物医药"真"创新政策鼓励下,国企该如何利用好政策优势聚集创新资源要素,Biotech公司应该如何与国企合作将创新资源要素转化成加速创新药落地的动力,解决当前行业寒冬下融资难、运营成本高的问题。
▷12:00-13:30
午餐
▷13:30-15:30
自由交流:国企和Biotech如何构架价值闭环实现共同增长及项目合作机会探讨
新时代,国企如何起到排头兵作用,如何做好自己,如何构建合作实现价值闭环模型,如何实现投资回报,如何激发国企的创新潜力;Biotech公司如何借助国企在资本、生产、政策、流通等要素的优势加速创新从实验室走向市场,提升国际技术竞争力。
▷15:30-15:50
论坛总结发言
蔡学钧 前BMS,全球研发副总裁
立即扫码报名
以临床转化为导向的临床前药效评价技术与经验分享
活动主办:格林泰科&药融圈 协办:BioBAY&艾名医学B馆-B106 8月16日
▼下滑浏览全部
▷09:00-09:05
主办方开场
刘婷格林泰科,市场部
▷09:05-09:30
体外药效活性筛选技术在新药成药性研究和临床转化的作用
杨晓东格林泰科,新药筛选平台负责人
▷09:30-09:55
类器官模型助力新药研发
周轶艾名医学,首席运营官
▷09:55-10:20
镇痛药物的开发现状及临床前药效评价方法
石刚格林泰科副总经理/市场项目部总监
▷10:20-10:45
肿瘤治疗性疫苗非临床安全性评价策略
夏玉叶苏州华测,机构负责人/副总经理
▷10:45-11:10
心血管系统疾病药物开发的动物模型选择
孙艳格林泰科,高级项目经理
▷11:10-11:35
帕金森药物的研发现状及临床前研究案例
江万祥格林泰科,副总经理/机构负责人
▷11:35-12:00
医疗器械的大动物有效性和安全性评价经验分享
李扶慧 格林泰科,医疗器械有效性评价负责人
立即扫码报名
天工所专场对接会
B馆-107 8月15日
▷13:30-13:35
中国科学院天津工生所领导致辞、药融圈致辞
▷13:35-13:50
中国科学院天津工生所整体情况介绍
▷13:50-14:20
研究所生物医药相关团队科研进展及成果介绍
▷14:20-15:00
参会企业介绍各自企业情况以及技术需求
▷15:00-15:30
座谈交流
立即扫码报名
江南大学合成生物学专场报告
B馆-中心舞台 8月15日AM
▼下滑浏览全部
▷09:40-09:45
领导致辞
▷09:45-10:00
功能化学品生物合成体系的创制与应用
聂尧 江南大学生物工程学院副院长
▷10:00-10:15
医药用酶/蛋白和高值氨基酸(衍生物)的合成生物制造技术
罗玮 江南大学生物工程学院教授
▷10:15-10:30
增强抗体免疫效应功能创新技术
吴志猛 江南大学生物工程学院教授
▷10:30-10:45
超强细胞修复蛋白-超分子弹性蛋白的合成生物设计与制造
吴俊俊 江南大学教授
▷10:45-11:00
酶催化非天然模式反应技术
赵群 江南大学教授
▷11:00-11:15
构建微生物细胞工厂高效合成大宗化学品和精细化学品
周丽 江南大学生物工程学院副教授
▷11:15-11:30
唾液酸化母乳寡糖的高效合成策略及应用
张洪涛 江南大学生物工程学院副教授
立即扫码报名
中国药科大学苏州校友会
中心舞台 8月15日
▼下滑浏览全部
主持人:张新悦,中国药科大学苏州校友会副秘书长/智银资本项目总监
▷ 14:00-14:05
开场致辞
胡珍泽
中国药科大学苏州校友会常务副会长/臻视特管理咨询创始人
▷14:05-14:10
开场致辞
蒲开富
中国药科大学苏州校友会秘书长/多加传媒总经理
苏州校友会企业合作需求发布
▷14:10-14:25
合作需求发布
全丹毅 江苏集萃新型药物制剂技术研究所,董事长、所长
▷14:25-14:40
药械组合产品包装验证和货架有效期验证
韩新荣 上海柯西医药科技创始人/浙江自贸区荣昇医药联合创始人
▷14:40-14:55
基因编辑猪低免疫原性生物材料开发及产业化
徐睿 成都中科奥格生物,CEO
▷14:55-15:10
聚焦肾科疾病领域产品开发及产业化合作
刘露 上海药坦, 商务总监
苏州校友会校友创业分享
▷15:10-15:30
苏州校友会校友创业分享
谢生荣 上海药坦, 商务总监
▷15:30-15:50
苏州校友会校友创业分享
田生容 苏州黛宜菲生物,创始人
▷15:50-16:50
Social Time:校友互动
立即扫码报名
科技创新与无创医美新蓝海论坛
B馆·中心舞台 8月16日
▼下滑浏览全部
▷09:00-09:10
加强美妆产品的合规化建设和质量监管
毛振宾中国食品药品质量安全促进会会长,中国药监局市场督察司原司长
▷09:10-09:25
无创美容的临床意义
郑志忠教授复旦大学附属华山医院教授
▷09:25-09:40
Overview of Microneedles
Sebastien HenryMicron Biomedical联合创始人Head of Technical Operations)
▷09:40-10:10
论坛:CMC of microneedles and its regulatory pathway
主持人:宇文镐原美国药监局高级审查官,现斯丹姆高级副总裁兼全球首席商务官CBO
嘉宾:
郑海安美国Albany药学院教授
杜 新 美国FDA药品审查官
徐百博士 纳通生物董事长
▷10:10-10:25
纳晶在治疗黄褐斑中的临床应用
骆丹教授江苏省人民医院教授
▷10:25-10:45
纳晶水光在皮肤美容中的临床应用
刘红梅院长 梅颜医美创始人
▷10:45-11:30
论坛:微针在临床应用中的评价方法和合规化使用
主持人:徐百博士纳通生物董事长
嘉宾:
郑志忠 复旦大学附属华山医院教授
刘红梅 梅颜医美创始人
宫 建 沈阳药科大学教授
童友之 开拓药业创始人 董事长
周 贵 北京智赢惠众医疗科技创始人
张庆瑞 苏州京东方医院
王恩波 苏州京东方医院
▷11:30-13:00
午休
▷13:00-13:15
万亿新蓝海,千亿大机会—医美行业品牌市场破局的思维及案例分享
刘逸春 念智品牌管理创始人
▷13:15-13:30
国家强监管是美容产业科技创新最大的战略机遇
田亚华教授全国美容医学教育资源共享联盟主席;中国整形美容协会新闻中心主任
▷13:30-13:45
开胃酒成交法在医美中的应用案例
李雪松极北光管理咨询的管理合伙人
▷13:45-14:15
论坛:白牌、货牌、品牌,泛医美行业品牌化之路的探索
主持人:徐百博士纳通生物董事长
嘉宾:
田亚华 全国美容医学教育资源共享联盟主席
刘逸春 念智品牌管理创始人
郑 岩 名相品牌管理(上海)有限公司 联合创始人
李雪松 极北光管理咨询的管理合伙人
方 承 北京双诚永道科技有限公司创始人
楚尚霖 米环文化联合创始人
▷14:15-14:30
如何应对FDA MoCRA法规对化妆品的监管
宇文镐 原美国药监局高级审查官,现斯丹姆高级副总裁兼全球首席商务官CBO
▷14:30-14:45
待定
Frank Roesken
Element Medical Aesthetics
▷14:45-15:15
论坛:生物医药与半导体前沿技术在美容行业的应用
主持人:许可纳米创始人
嘉宾:
徐 弢 深圳清华研究院生物智能制造和活体打印中心主任
邢海英 湃肽生物董事长
宗东升 沈阳药科大学 医疗器械学院产学研用融合促进中心 执行主任
许 可 苏州唯思尔康科技有限公司 任董事长CEO
姬胜利 润辉生物技术(威海)有限公司 董事长
潘 敏 纳通生物 纳晶首席体验官
▷15:15-15:45
论坛:消费医疗的投资机会
主持人:郝格
嘉宾:
万 钊 御海资本
胡 斌 斌哥医械圈
杨可逸 磐缔创投合伙人
林 雷 建银国际医疗成长基金董事长
▷15:45-16:30
大赛:大学生绿色化妆品创新创业大赛优胜作品汇报
主持人: 徐百博士 纳通生物董事长
评委:
殷建国 生物纳米园董事长
万 钊 御海资本
程 伟 中国抗衰老促进会化妆品产业分会会长
胡 斌 斌哥医械圈
郭思周 时节创投
邓建良 天汇资本
林 雷 建银国际医疗成长基金董事长
杨可逸 磐缔创投合伙人
高 垒 深圳精宬科技有限公司 CEO孔 雷 千行资本合伙人
于立燕 苏州工业园区管委会科技招商中心
郑叶萍 无锡锡山开发区
陈勇奇 江阴国家高新区
▷16:30-18:15
参观纳晶研发生产车间
▷18:15-21:30
晚宴
立即扫码报名
合成生物学产业化难点
主办单位:ATS安拓思&药融圈
B馆-D04 8月15日 09:15-12:15
▼下滑浏览全部
8月15日
▷ 09:15-09:30
开场评弹表演
▷09:30-10:15
话题一:合成生物与保健品
1.合成生物学原料市场2032年将突破100亿美元? 如何打通产业化道路?
2.虾青素、角鲨烯、NMN、HMOS(母乳低聚糖)......合成生物学选品如何综合技术与市场?
3.保健品成分竞争下的赛道选择:开发新产品,还是迭代旧技术?
嘉宾
张宝琪 百开盛生物总经理助理
张 琦 邦泰生物创始人兼CSO
方 诩 恒鲁生物创始人兼首席科学家
刘振云 一兮生物 创始人兼CEO
▷10:15-10:30
抽奖互动
▷10:30-11:15
话题二:合成生物与医学
1.GLP-1药物开发难点与生产挑战。
2.合成生物学开发多肽类药物的成本优势。
3.生物催化在医药领域的应用痛点及解决方案。
嘉宾
张 仙 百福安生物副总裁
朱 毅 杭州珲信生物副总裁
黄科学 华润双鹤合成生物研究院院长,首席科学家
▷11:15-11:30
抽奖互动
▷11:30-12:15
话题三:合成生物与医美
1.产业化中,合成生物学方法合成胶原蛋白/透明质酸的优势。
2.风口还是红海?三因赛道的竞争与难点。
3.化妆品活性原料行业创新的“新引擎”:合成生物学如何布局下游应用研究。
嘉宾
张 骊元一生物创始人/CEO
赵 俊合成生物蛋白安徽省联合共建学科重点实验室主任
丁 威仅三生物创始人
周孟博宁波糖聚新材料创始人
立即扫码报名
多肽药物开发论坛——挑战与创新
主办单位:甘肃省百事兴药业&药融圈
B馆-E04展位圆桌对话区 8月15日 10:30-11:10
▼下滑浏览全部
圆桌对话会:多肽药物开发论坛—挑战与创新
话题:
1.如何延长多肽药物半衰期?
2.药物专利到期后,多肽原料药企业如何抓住机遇?
3.多肽药物创新可以从哪些方面入手?
4.多肽药物的生产上的难点如何避免?
5.多肽药物的商业化落地如何做?
拟邀嘉宾
诸 旭江苏普莱生物 首席科学官
刘志国湃肽生物,联合创始人
汪兆伟百事兴科技 董事长
丁 伟南京知和医药,副总经理、首席科学家
庞 力恒敬合创生物,联合创始人、CFO
付玉清健元医药,高级研发总监
* 嘉宾排名不分先后,更多嘉宾持续更新中...
立即扫码报名
国产培养基产业发展研讨会
主办单位:甘肃省百事兴药业&药融圈
B馆-E04展位圆桌对话区 8月15日 14:00-14:40
▼下滑浏览全部
圆桌对话会:国产培养基产业发展研讨会
话题:
1.高性能培养基如何助力药物研发?
2.国产培养基厂家如何改善批量生产稳定性问题?
3.药企对培养基的使用粘性大,国产厂家如何占领市场?
4.选择合适的培养基,需要关注哪些问题?
拟邀嘉宾
李明新思鹏生物 CEO
刘旭平倍谙基生物 总裁
刘翠华百奥泰生物 高级副总裁
韩向宗依科赛生物,研发副总裁
汪兆伟百事兴科技 董事长
* 嘉宾排名不分先后,更多嘉宾持续更新中...
立即扫码报名
冰火碰撞,百诚医药今夏再邀您咖啡共谈,相聚姑苏
主办单位:百诚医药&药融圈
B-F07 8月15日-16日
现场冰爽咖啡与热点话题共撞
• 产品转移过程的关注点
• 呼吸领域特色仿制和改良新推荐
• 百诚可转让进度优势项目集锦
• 经典名方开发的立项考虑与推荐
• 助力创新药研发的非临床药理和生物评价技术
• 国内CXO行业的增长点—中药CXO
立即扫码报名
C馆-药学CMC&创新&CXO馆
新药创始人峰会
C馆-C102 8月15日
▼下滑浏览全部
主持人:邓永奇 凯复医药,CEO
▷09:00-09:25
如何应对复杂的生物医药产业环境(拟)
王兴利 复星医药,执行总裁&创新药事业部联席CEO
▷09:25-09:50
华润医药在生物医药领域打造新质生产力的探索和思考
崔兴品华润医药集团,副总裁、华润三九医药董事
▷09:50-10:15
绕不开的商业化,Biotech公司该怎么看待商业化?
刘 谦 阿斯利康中国,副总裁
▷10:15-10:40
当前市场环境下生物医药初创公司的发展策略
刘为民 百济神州广州创新中心生科创投,董事长总经理
▷10:40-11:05
中国生物医药产业投资机会分析
毛 化 沙利文 ,大中华区合伙人兼董事总经理
▷11:05-12:00
圆桌:中国Biotech公司未来突破点有哪些?
1.被MNC并购,前提是准备好没?
2.进入集采,创新药的想象力真的那么好?
3.被国内大药企并购退出?
4.抓住机会从Biotech到Biopharm?
5.以仿养创?怎么养?
6.兼顾CRO业务获得现金流?卷得动吗?
邓永奇 凯复医药,CEO(主持人)
刘为民 百济神州广州创新中心生科创投,董事长总经理
金 方 健康元,首席科学家
刘 谦 阿斯利康中国,副总裁
吴世斌 万邦医药,董事长秦锁富 科兴制药,VP&研究院院长
▷12:00-13:30
午餐
中国新药的理性发展方向
策划人&主持人:党群 真实生物,总裁
▷13:30-13:55
创新药是选FIC还是BIC?解决临床未满足需要是关键!
党
群真实生物,总裁
▷13:55-14:20
和誉医药FGFR抑制剂开发进展
陈
椎 和誉医药,联合创始人兼CSO
▷14:20-14:45
CAR-T治疗实体瘤的屏障及研究实践
章登吉 美迪西,生物技术药物分析部负责人
▷14:45-15:10
YAP/TEAD抑制剂的开发
朱继东 奕拓医药,CEO
▷15:10-15:35
浅谈“前药”的前世今生-长乘医药的前药研发
宋钦辉 长乘医药,CMO
▷15:35-16:00
慢乙肝功能性治愈创新药物的研究与开发
张
弛西安新通药物,副总经理
▷16:00-16:25
Delivery
of Excellence - 递送引导的药物创新
沈
旺维眸生物,创始人、董事长兼CEO
立即扫码报名
FDA对新兴医疗产品的监管联合主办:FDA专家协会
C馆-C101 8月15日
▼下滑浏览全部
主持人:李宁,君实生物,副董事长
▷09:00-09:05
致辞李 宁 君实生物,副董事长
▷09:05-09:20
新兴医疗产品的概述杜 新 埃格林医药,联合创始人&CEO
▷09:20-09:45
新兴医疗产品的发展趋势和监管挑战陆金华 星尘生物,CSO
▷09:45-10:10
CGT产品临床试验的注意要点门宇欣 海昶生物,首席医学官
▷10:10-10:35
CGT产品的临床药理学考量杨 劲 中国药科大学,教授
▷10:35-11:00
CGT产品的cGMP合规考量孙志刚 绿叶制药集团,高级副总裁
▷11:00-11:25
cGMP合规考量下除菌过滤验证的挑战
杨潇军 挪亚泰尔,技术负责人
▷11:25-12:10 小组讨论:新兴医疗产品的国际化
李 宁 君实生物,副董事长
杜 新 埃格林医药,联合创始人&CEO
张明东 苏州赛纳思医疗,CMO
王幼民 银泉生物制药咨询有限公司,创始人&首席顾问
孙 鹤 天士力控股集团,全球副总裁
胡育志 香港白橡树,CEO
▷12:10-13:30
午餐
主持人:陈少羽, 美国安诺波特律师事务所驻上海代表处,管理合伙人
▷13:30-14:00
小核酸药物的开发与监管考量杨永胜 海昶生物,高级副总裁&核酸创新研究院院长
▷14:00-14:30
FDA对溶瘤病毒产品的的监管张 静 杰科生物,CMO
▷14:30-15:00
首个DMD基因疗法的FDA审评审批争议-案例研究王亚宁 瑞宁康生物医药,创始人&CEO
▷15:00-15:30
美国食品药品监督管理局在药品研发中使用人工智能和机器学习的监管概况韩晓梅 E7 CAPITAL创始人&CEO
▷15:30-16:00
人工智能/机器学习创新医疗器械监管策略胡云富 泛生子,首席医疗官
▷16:00-16:45
小组讨论:新兴医疗产品监管的进一步深化
陈少羽 美国安诺波特律师事务所驻上海代表处,管理合伙人
陆金华 星尘生物,CSO
韩晓梅 E7 CAPITAL,创始人&CEO
窦金辉 挚盟医药,注册法规事务副总裁
王亚平 蓝鹊生物,首席医学官
立即扫码报名
临床开发论坛大会主席:杨修诰,石药集团,高级医学总监 郑 航,重庆医科大学,教授
C馆-C103 8月15-16日
▼下滑浏览全部
8月15日
主持人:孟杰天,安徽万邦,首席医学官
▷09:00-09:25
从临床角度看中枢神经系统药物的临床开发策略及案例分享
申华琼 纽欧申医药,创始人&CEO
▷09:25-09:50
新药临床研发的适应症选择策略
朱永红 岸迈生物,CMO
▷09:50-10:15
关键临床试验失败案例分析
陈
霞 泰格医药,高级副总裁&首席医学官
▷10:15-10:40
抗体偶联药物(ADCs)的剂量优化相关临床研究及安全管理措施分析
杨修诰 石药集团,高级医学总监
▷10:40-11:05
从临床角度看ADC开发的挑战和前景
石
燕启德医药,首席医学官
▷11:05-11:30
中国制造,出海有多难?
杜一鸣 上海海和药物,高级副总裁
▷11:30-12:10
圆桌:中国创新药注册及临床全球化操作技术的短板?
杨修诰 石药集团,高级医学总监刘艳玮 武田中国,副总裁 ,武田大中华区注册事务部负责人
杜一鸣 上海海和药物,高级副总裁
石 燕 启德医药,首席医学官
▷12:10-13:30
午餐
论坛主持人
苏泉宇 一临云,联合创始人&首席商务官
▷13:30-13:55
从临床视角看抗肿瘤创新药的临床开发策略及案例分享
隋
红浙江瀛康生物医药有限公司,医学总监
▷13:55-14:20
神经退行性疾病药物发展:从过去看未来
陈柏州 加立生科,CEO
▷14:20-14:45
新药研发早期的统计方法
赵 萌苏州信诺维,统计及编程执行总监
▷14:45-15:10
司美格鲁肽临床开发策略及要点
孟杰天 安徽万邦,首席医学官(CMO)
▷15:10-15:35
以终为始-数据分析驱动,提高创新药临床开发成功率
戴鲁燕 粹羽咨询,创始人
8月16日 临床试验创新设计与高质量发展
主持人:郑航,重庆医科大学药学院,教授
▷09:00-09:30
新质生产力引领临床试验高质量发展
周 焕 中国药理学会药物临床试验专委会青年主任委员
▷09:30-09:55
临床试验数字化的现状与未来
郑 航 重庆医科大学药学院,教授
▷09:55-10:20
基因治疗产品的早期临床研究及案例分享
吕 华 天泽云泰,高级副总裁
▷10:20-10:45
临床研究创新设计类型与统计实施
汤在详 苏州大学,教授
▷10:45-11:10
抗肿瘤药物临床试验中的终点选择
任以中 葆元医药,医学事务高级总监
▷11:10-11:30
中国临床试验出海的机遇与挑战
邓晓宇 希毅医学,创始人/总经理
▷11:30-11:50
真实世界数据在临床研究中的应用
胡 皓 医数康成,总经理
▷11:50-13:30
午餐
▷13:30-14:00
监管科学视角下临床研究设计和质量的重大意义
侯 艳 北京大学,生物统计系研究员
▷14:00-14:30
我国药械组合产品监管新进展
许 伟 国家药品监督管理局医疗器械技术审评中心原副主任
▷14:30-15:00
数字化助力监管决策——面向药品现代化监管的智能化服务平台
王维玉 北京灵迅医药科技公司,联合创始人
▷15:00-15:20
从患者角度出发看DCT临床实践的融合与落地
夏素琴 创达医药,总经理
▷15:20-15:40
临床试验的“质”与“量”的博弈 — 付出越大,环节越多,质量就越好吗?
张 淼 南京麦普斯医药科技有限公司,创始人
▷15:40-16:00
临床试验不良事件的损害赔偿与风险管理
刘亚卿 上海浦东五新保险经纪有限公司,副总经理
立即扫码报名
免疫治疗新药开发论坛
C馆-C101 8月16日
▼下滑浏览全部
策划人&主持人:张立刚,冠科生物,中国商务区总经理
▷09:00-09:20
转化医学研究协助HX009 CD47/PD-1双功能抗体的临床开发
李其翔 翰思生物,联合创始人&总裁和首席科学官
▷09:20-09:40
免疫疗法:双抗vs细胞治疗
顾津明 传奇生物,前中国区研发负责人
▷09:40-10:00
ImmunoCytokine: The New Era of Cancer Immunotherapy
殷刘松 盛禾生物,CEO&CSO
▷10:00-10:20
Regulatory T Cells and Personalized Immunotherapy
李斌上海交通大学,特聘教授、上海市免疫学研究所科研副所长
▷10:20-10:40
TBD
卢宏韬 科望医药,联合创始人兼首席科学官
▷10:40-11:00
动物模型在I/O药物开发中的应用
王晶晶 冠科生物,太仓公司总经理
▷11:00-11:40
圆桌对话:从2024国际会议看全球免疫疗法治疗趋势和机会
李 佳 朗盛投资,合伙人(主持人)
李 丹 博亚方洲创投,副总经理
易 琳 德福资本,合伙人
时永灵 聚明创投,合伙人
宁高见 元生创投,董事总经理
▷11:40-13:30
午餐
▷13:30-13:50
JAK1抑制剂在自免领域的应用
姜 非 康哲药业,大中华区首席投资官
▷13:50-14:10
双特异性抗体药物项目的开发进展
吴辰冰 岸迈生物,CEO
▷14:10-14:30
用于肿瘤免疫治疗的新型腺苷受体A2aR/A2bR拮抗剂的发现
王永辉 复旦大学讲座教授、励缔医药联合创始人
▷14:30-14:50
异体CAR-T药物开发进展
殷文劼 亘喜生物,药理毒理部高级总监
▷14:50-15:10
应用肿瘤类器官技术发现临床候选化合物
王 鹏 冠科生物,体外研发执行总监
▷15:10-15:30
胰腺癌个性化多肽
陈海敏 安达生物,COO
立即扫码报名
抄底并购之年:中国Biotech的新出路,新活法、新征程
C馆-C102 8月16日
▼下滑浏览全部
策划人&主持人:陈锋 科林利康副总裁/博羚资本负责人
▷09:00-09:05
联合主办方致辞
陈锋 科林利康副总裁/博羚资本负责人
▷09:05-09:35
主题演讲:2024全球生物医药License/并购趋势与策略
胡奇聪 波士顿咨询(BCG)合伙人、董事总经理
▷09:35-10:45
圆桌讨论:MNC如何与中国药企创新合作与双向奔赴
夏明德英诺湖医药创始人、CEO(主持人)
陈 锋科林利康副总裁/博羚资本负责人
王 昕施维雅全球拓展亚太BD总监
▷10:45-12:00
圆桌讨论:穿越寒冬,中国本土Biotech的BD策略思考与交易前景分析
董 丹 济民可信,业务发展负责人(Moderate)(主持人)
邹丽晖 达歌生物CEO、联合创始人
胡会国 迈威生物董事、首席商务合作官、高级副总裁、董秘
姜 非 康哲药业首席投资官(大中华区)
杨建国 近邻生物CEO
▷12:00-13:30
午餐
▷13:30-13:50
创新路演1
王 喆 长森药业,创始人
▷13:50-14:10
创新抗板药物,提升心脑血管病患者治疗药效和安全性
陈小五 柯君医药化学副总裁
▷14:10-14:30
专注于细胞和基因治疗领域的生物研发赋能平台
夏珏妤 拓弘康恒,副总经理&首席战略发展官
▷14:30-14:50
为全球抗生素耐药问题提供差异化解决方案
邓一军 MetCura,创始人&CSO
▷14:50-15:10
创新路演5
张金晶 威斯津生物,商务发展总监
▷15:10-15:30
创新路演6
张 弛 西安新通药物,副总经理
▷15:30-15:50
NuRapid® 新一代分子诊断技术产业化平台
何林富 为真生物,副总裁、CTO
▷15:50-16:10
创新路演8
邢 莉 英伟达,创始人/CEO
▷16:10-16:30
全球首个基于BCMA/CD19双靶并联的CAR-T细胞疗法的研发进展
蔡祥海 科弈药业,研发副总裁
▷16:30-16:50
创新路演10
魏寅祥 永心生物,项目总监
立即扫码报名
小分子药物开发策略
C馆-C104 8月15日
▼下滑浏览全部
主持人:李丽艳,默克中国,数字化产品经理
▷09:00-09:20
一种新型的基于分子胶payload的小分子偶联药物开发 任志华 开拓药业,VP/新药研究院院长
▷09:20-09:40
无人实验室技术及全新的化学实验工作模式
乔 木 云合成,总经理
▷09:40-10:00
AI加速化学合成的趋势李丽艳 默克中国,数字化产品经理
▷10:00-10:20
中国创新药的困境、挑战和转型胡邵京 思康睿奇,CEO
▷10:20-10:40
AI+自动化赋能新药研发徐 涛 智化科技,副总裁
▷10:40-11:00
利用晶型技术评估和提高化合物的可开发性徐 俊 晶云药物,研发高级总监
▷11:00-11:20
AI计算驱动的变构小分子激动剂开发沈倩诚 宇道生物,CEO
▷11:20-13:30
午餐
▷13:30-13:50
Rediscovery of Paclitaxel: An innovative oral Paclitaxel softgel capsule product for improving cancer therapeutics黄慧瑜 美济生物,首席科学官
▷13:50-14:10
怎么做好创新药研发风险评估?陈 洪 外专局特聘专家
▷14:10-14:30
痕量杂质研究案例分享与高难度杂质研究平台冯胜昔 艾奇西,总经理
▷14:30-14:50
神经系统疾病药物临床前药理研究要点江万祥 格林泰科,副总经理/机构负责人
▷14:50-15:10
药物可开发性评估和处方前研究唐开勇 泓博智源,副总裁
▷15:10-15:30
亚硝胺药物相关杂质的风险评估和控制策略考量曹煜东 联拓生物,CMC 负责人
▷15:30-15:50
消化道疾病及消化道肿瘤免疫创新药开发胡平生 生诺生物,CEO
立即扫码报名
高校科研院所生物医药转化论坛
论坛大会主席&主持人:罗有福,四川大学,教授
论坛副主席:叶庭洪,四川大学,教授
论坛秘书长:欧阳亮,四川大学,教授
C馆-C104 8月16日
▼下滑浏览全部
主持人:罗有福,四川大学,教授
▷09:00-09:25
细菌耐药性与抗菌药物研发杨玉社 上海药物研究所
▷09:25-09:50
靶向线粒体ClpP的创新药物研发罗有福 四川大学,教授
▷09:50-10:15
基于未满足的临床需求如何优化新药分子设计钱 海 中国药科大学,理学院院长
▷10:15-10:40
靶向RNA可变剪接抗肿瘤药物研发张红河 浙江大学,教授
▷10:40-11:05
神经退行性疾病新靶点和先导小分子发现欧阳亮 四川大学,教授
▷11:05-11:30
蛋白激酶抑制剂研究:强弩之末,还是正当其时?张 健 复旦大学,教授
▷11:30-11:55
抗纤维化药物转化研究叶庭洪 四川大学,博士生导师
▷11:55-13:30
午餐
▷13:30-13:55
靶向电子传递链QcrB以及直接靶向InhA的抗分枝杆菌新药研发张天宇 中国科学院广州生物医药与健康研究院(GIBH),感染与免疫研究中心副主任
▷13:55-14:20
抗耐药真菌新技术、新靶点和新先导物的发现及转化研究张大志 海军军医大学,教授
▷14:20-14:45
靶向蛋白降解药物的基础研究和转化董晓武 浙江大学,教授
▷14:45-15:10
聚焦合成致死2.0策略,为MTAP缺失的肿瘤提供全方位的创新解决方案陈旭星 优理生物,创始人&首席执行官
▷15:10-15:35
巨噬细胞干预纳米药物用于消化道肿瘤治疗刘祥瑞 浙江大学医学院,教授/奥睿德医药创始人
▷15:35-16:00
基于脑肠轴的多发性硬化发病机制研究与干预周嘉伟 中国科学院脑科学与智能技术卓越创新中心/神经科学研究所,研究员
立即扫码报名
创新药中美双报如何制定研发策略
C馆-C105 8月15日
论坛策划人&主持人
邹灵龙 康维讯生物,创始人、董事长&CEO
▼下滑浏览全部
▷09:00-09:30
创新TCE技术降低CRS风险提升成药潜力曹国帅 天港医诺,研发负责人
▷09:30-10:00
Strategies for accelerating the time and reducing the cost of orphan drug global development in gene therapyAlvin Luk 辉大基因,联合创始人&首席执行官
▷10:00-10:30
中美IND申请非临床研究要点贺全仁 艾博生物,药理毒理高级副总裁
▷10:30-11:00
中美申报CMC要点Audrey Jia Wonderfulland LLC,CEO,前FDA CMC审评员
▷11:00-11:30
中美双报生物分析策略与实操分享邹灵龙 康维讯生物,创始人、董事长&CEO
▷11:30-13:30
午餐
▷13:30-14:00
Immunogenicity Testing Strategy & Regulatory Expectation on Method Validation- FDA perspective严皓珩 复宏汉霖,美国注册负责人,前FDA评审
▷14:00-14:30
出海申报的临床试验设计与数据管理杨修诰 石药集团,高级医学总监
▷14:30-15:00
支持出海的ADC毒素ADME研究朱明社 锐迪欧医药,首席科学官&首席顾问
▷15:00-15:30
中美药品电子申报 (eCTD)要点任 翀 复宏汉霖,药政注册副总监
立即扫码报名
制剂出海论坛
C馆-C105 8月16日
▼下滑浏览全部
▷09:30-10:00
乘风破浪:中国医药企业出海现状与展望骆世忠 岸金”医健国际化“创新合作中心创始合伙人及岸金生物CEO
▷10:00-10:30
仿制药出口美国市场的实践与思考钭勇亮美国吉马莱博有限公司中国代表处首席代表
▷10:30-11:00
扬帆远航:制剂出海助力企业长期发展安晓霞上海迪赛诺医药集团股份有限公司总经理&董事长
▷11:00-11:30
制剂出海欧洲的机遇和问题关注
荆士恒汇宇制药英国海玥分公司及欧洲事业部总经理
▷14:00-14:30
海外临床试验之项目管理王丽娜知名药企临床运营高级顾问
▷14:30-15:00
满足临床需求,提高治疗可及性,论FDA 505b2创新药研发孙 鹤天士力控股集团全球副总裁
▷15:00-15:30
医药企业出海和BD交易架构设计及法律关注要点刘婷婷上海市锦天城律师事务所资深律师
立即扫码报名
纳米&复杂注射剂大会
C馆-C106 8月15-16日
▼下滑浏览全部
8月15日
精准递送-纳米制剂专场
▷9:30-10:00
克服递药屏障纳米药物制剂研究进展及展望陆伟跃复旦大学教授
▷10:00-10:30
纳米药物的设计与临床转化申有青
浙江大学教授
▷10:30-11:00
纳米脂质注射剂及其用于药物递送的研究和产业化
张馨欣
中国科学院上海药物研究所研究员
▷11:00-11:30
纳米结晶可控制备与体内命运的研究卢懿 复旦大学副教授
▷14:00-14:40
细胞毒类药物纳米制剂的产业化新技术开发及案例分析
曹青日
苏州大学教授
▷14:40-15:20
基于天然化合物的新型多功能肿瘤靶向脂质体研发王建新复旦大学教授
▷15:20-16:00
纳米技术促进药物皮肤吸收与储留
吕慧侠
中国药科大学教授
▷16:00-16:40
纳米乳剂的研发实践和思考
袁旭东艾康药业创始人&CEO
8月16日
攻克高精尖-复杂注射剂专场
▷9:30-10:00
高端制剂-长效注射剂的现状、进展和思考
李亚平
中国科学院上海药物所研究员
▷10:00-10:30
复杂纳米注射剂CMC中的表征研究周金鹏浙江智达药业有限公司研发总监中国药科大学教授
▷10:30-11:00
可吸收聚合物在长效注射给药系统中的应用
栾瀚森
璟熙医药科技(深圳)有限公司高级技术顾问
▷11:00-11:30
复杂注射剂从小试、中试、到大生产应该注意的工艺问题
徐上虎
四川汇宇制药化学研究二室主任
▷11:30-12:00
复杂注射剂逆向工程研究分享
李文博
石家庄凯瑞德医药科技发展有限公司 总经理
▷14:00-14:30
原位凝胶在药物递送中的应用和难点祁小乐 中国药科大学副教授
▷14:30-15:00
PIC/S GMP无菌药品附录对我国无菌药品检查的影响
夏禄华
重庆誉颜制药有限公司副总裁
▷15:00-15:30
复杂注射剂产品设计与质控策略
周建平
中国药科大学教授
立即扫码报名
创业者的思考——新质生产力驱动生物医药未来之路
主办单位:云合成&融域智慧&药融圈
C馆-F07 8月15日 13:30-15:20
▼下滑浏览全部
▷13:30-14:15
话题一:国内药物研发智能化、规范化破局之路
1.创业者思考:面对内卷,生物医药企业如何“练内功”突破困局?
2.生物医药产业化痛点怎么破:研发力、生产力、市场化...
3.从数据到管理,如何高效管理研发数据有效性、安全性、准确性、完整性
徐 俊正济药业 创始人、董事长兼总裁
李洪明广生堂药业 首席运营官
刘怀振川成医药 董事长
何建国重庆南松凯博生物 董事长
彭学东威胜生物医药联合创始人
徐红岩吉尔生化 董事长
袁永坤苏州亚科科技股份 总经理
▷14:15-14:25
沉浸式VR交互体验
▷14:25-15:10
话题二:国内CXO高效化、合规化破局之路
1.CXO为主的医药领域,如何探索符合企业发展需求的“新质生产力”
2.生物医药研发生产无人化质量合规探讨
3.生命科学领域实验室如何实现降本增效
陈 恬车头制药 董事长
王 雷都创医药 CEO
赵 磊青木制药 总经理
赵小林南京哈柏医药 总经理
彭立军上海皓元医药 副总裁/首席商务官
▷15:10-15:20
抽奖互动
立即扫码报名
D馆-MAH&CXO&DDS馆
MAH B证企业高峰论坛联合主办:乐陵市循环经济示范园
D馆-D101
▼下滑浏览全部
8月15日上午入门篇—B证企业法规政策主持人:卜华荣 MAH专家
▷09:30-10:00
B证企业关键人员能力素养要求谢海燕 丽珠医药集团质量管理总部总经理兼制剂质量总监;丽珠医药集团B证质量负责人;广东省药学会 药品生产质量受权人专业委员会 主任委员
▷10:00-10:30
质量体系生命周期管理—质量成熟度(QLM--QMM)毕瑞凤 资深GMP专家
▷10:30-10:50
MAH产业发展及乐陵市循环经济示范园推介
▷10:50-11:30
强监管下B证企业如何提升质量管理体系卜华荣 MAH专家
▷11:30-12:00
药品专利纠纷早期解决机制朱瑞 北京精金石知识产权代理有限公司专利咨询师
8月15日下午生产篇—B证企业委托生产
▷14:00-14:30
药品委托生产现状与监管考量邵蓉 中国药科大学NDPE执行副主任、药品监管科学研究院执行院长
▷14:30-14:50
MAH药品全生命周期管理实践经验刘超 山东百诺医药股份有限公司市场部总监
▷14:50-15:20
B证企业委托生产共线风险评估及案例分享谭宏宇 国家资深GMP检查员、原CFDA骨干检查员
▷15:20-15:40
药品上市许可持有人委托生产质量体系建立要点及风险分析张法语 北京康利华咨询服务有限公司+项目经理、高级GMP咨询师
▷15:40-16:10
MAH制度和未来趋势
鲍鹏九州通/京丰集团副总经理
8月16日上午产品篇—B证企业产品立项
▷9:30-10:00
集采下半场企业产品线调整策略与仿制药剩下的选项机会
王立峰慧药咨询创始人
▷10:00-10:30
MAH制度下药品工艺技术管理吴军 药品制造系统工程与GMP专家
▷10:30-11:00
MAH制度下产品技术转移法规要求及常见问题
李宏业 北京宏汇莱科技有限公司总经理兼总工程师
▷11:00-11:30
MAH B证企业药品注册现场核查关注点及缺陷案例分析杨老师 国家GMP检查员,国家药监局高级研修学院特约讲师
8月16日下午变更篇—B证企业变更管理
▷14:00-14:30
药品上市后变更研究与申报关注问题药监老师
▷14:30-15:00
MAH制度下场地变更法规要求和实操指南马海涛 广州国标医药科技有限公司 研发技术副总
▷15:00-15:30
MAH制度下持有人变更法规要求和实操指南丁恩峰 业内资深GMP、药政法规与制药技术专家
立即扫码报名
吸入制剂大会
D馆-D102
▼下滑浏览全部
8月15日上午百亿赛道—吸入制剂究竟该如何做?
▷9:30-10:00
对吸入给药创新的思考 游一中 常州市第一人民医院主任药师、副主任医师;中国包装联合会气雾剂专业委员会终身荣誉主任;中国颗粒学会吸入颗粒专委会名誉主任
▷10:00-10:30
中药吸入制剂研发的挑战与应对策略廖永红 中国医学科学院药用植物研究所 研究员
▷10:30-11:00
改良型吸入制剂开发策略与难点分析吴闻哲 医药先进制造国家工程研究中心 经鼻及吸入给药研究室负责人
▷11:00-11:30
储库式吸入粉雾剂开发难点及对策史楷岐 博士、苏州易合医药有限公司 创始人&总经理
8月15日下午乘风破浪—吸入制剂BE研究挑战性在哪?
▷14:00-14:30
吸入疗法在慢性气道疾病治疗中的地位与展望殷凯生 南京医科大学第一附属医院呼吸与危重症医学科二级教授、主任医师、博导;中华医学会呼吸分会哮喘学组顾问;中国哮喘联盟总负责人(之一)
▷14:30-15:00
吸入制剂人体生物等效评价研究郑恒 国家药品审评中心专家、 湖北省药监局核查专家
▷15:00-15:45
经口鼻吸入制剂关键质量属性研究以及吸入仿制药体外生等效APSD评价方法及重点 EMA FDA 指导原则的更新沈丹蕾 全国吸入给药联盟主席、南京白令信息科技有限公司总经理
▷15:45-16:05
吸入制剂临床试验质量关键点探讨周燕 安徽万邦医药副总(VP)
8月16日上午未来已来—吸入制剂产业化壁垒?
▷9:30-10:00
高壁垒吸入剂开发,如何占据竞争优势佟振博 东南大学教授
▷10:00-10:30
鼻喷雾剂的开发及质量研究谷丽 南京樟益医药科技有限公司 创始人&总经理
▷10:30-11:00
PulmoSol 与 PulmoSphere:干粉吸入剂制剂技术及研发难点刘恒利 凯信远达医药有限公司 研发中心负责人
▷11:00-11:30
吸入制剂包装相容性研究与实施蔡荣 高级工程师(教授级)、原上海市食品药品包装材料测试所
8月16日下午道阻且长—吸入制剂如何翻越审批大山?
▷14:00-15:00
简评欧美吸入制剂审评及指导原则张星一 原CDE高级审评员
▷15:00-15:45
干粉吸入制剂的中欧审评评价及技术难点陈永奇 珠海瑞思普利医药科技有限公司董事长兼首席科学家
立即扫码报名
透皮制剂大会
联合主办:华溶仪器
D馆-D103 8月15-16日
▼下滑浏览全部
8月15日上午主持人:马海涛 广州国标医药科技有限公司研发技术副总
▷9:30-10:00
透皮制剂产品开发的国内外法规对比及案例分析全丹毅 江苏集萃新型药物制剂技术研究所董事长、所长
▷10:00-10:30
智能微针递药器件:创新与转化张宇琪 浙江大学研究员
▷10:30-11:00
经皮半固体制剂的处方工艺开发和表征谷丽 南京樟益医药科技有限公司创始人、总经理
▷11:00-11:30
基于垂直扩散池法的IVPT在外用制剂研发中的应用段云剑 深圳市华溶分析仪器有限公司技术总监
8月15日下午主持人:朱亚东 苏州易科新创科学仪器有限公司技术应用总监
▷14:00-14:30
Innovations for health. TTS/OTF/MAP/OBDS drug delivery solutions for improved patients’ outcomeStefan Arnold LTS Lohmann Therapie-Systeme AG亚洲业务发展总监兼亚洲办事处主任
▷14:30-15:00
差异化高端经皮制剂的开发策略及案例分析
吴传斌 山东大学药剂学教授、药物制剂研究所所长
▷15:00-15:30
透皮微针制剂的开发与应用翟光喜 中国药科大学教授、博士生导师
▷15:30-16:00
TDDS改良型新药开发案例—从立项论证到临床研究张海龙 长沙晶易医药科技股份有限公司总裁
▷16:00-16:30
【圆桌论坛】透皮制剂开发领域挑战—原料、药学、临床、生产
主持人:朱亚东苏州易科新创科学仪器有限公司技术应用总监
盛晓霞 杭州领业医药科技有限公司 总经理
郭晓迪 浙江华海药业首席科学家兼制剂研究院院长
姜宏梁 武汉宏韧生物医药股份有限公司董事长
张海龙 长沙晶易医药科技股份有限公司总裁
肖稳定 湖南九典制药股份有限公司药物研究院院长兼湖南普道医药技术有限公司总经理
8月16日上午主持人:刘亚利 汕头大学医学院第一附属医院,机构副主任、I期病房主任
▷9:30-10:00
透皮制剂技术审评的逻辑体系与实践张星一 原CDE高级审评员
▷10:00-10:30
透皮产品的临床研究设计—实例分享刘亚利 汕头大学医学院第一附属医院,机构副主任、I期病房主任
▷10:30-11:00
经皮给药制剂临床试验的实验设计、实施、生物分析要点姜宏梁 武汉宏韧生物医药股份有限公司董事长
▷11:00-11:30
可溶微针技术在医药领域的应用和挑战 李成国 优微(珠海)生物科技有限公司联合创始人
8月16日下午
主持人:王永刚 世中联经皮给药专业委员会副会长,沈阳永晟康泰医药科技有限公司董事长/正高级工程师
▷14:00-14:30
透皮制剂的辅料现状与展望 吴正红 中国药科大学教授、博士生导师
▷14:30-15:00
经皮给药:现状、机会及策略
刘莱 北京湃驰泰克医药科技有限公司总经理
▷15:00-15:30
热熔胶贴膏发展概况及应用前景分析王永刚 世中联经皮给药专业委员会副会长,沈阳永晟康泰医药科技有限公司董事长/正高级工程师
立即扫码报名
对话仿制药决策层
D馆-D104
▼下滑浏览全部
8月15日
▷09:30-10:15
群雄逐鹿,仿制药企业转型路在何方?
耿鸿武 清华大学老科协医疗健康研究中心,执行副主任
▷10:15-12:00
核心观点碰撞 | 对话企业家/决策层
刘春波 南京恩泰医药科技有限公司 董事长郑忠辉 山东新华制药股份有限公司 副总经理唐建飞 杭州朱养心药业有限公司/浙江康润制药有限公司 总经理
吴四清 湖南九维生物医药有限公司 董事长
李晓祥 安徽省新星药物开发有限责任公司 董事长兼总经理
江 鸿 江西施美药业股份有限公司 创始人、董事长兼CEO;山东创新药物研发有限公司 董事长兼CEO
汤怀松 江苏华阳制药有限公司 董事长
胡仁军 武汉九州钰民医药科技有限公司 董事长 陈 磊 湖南中晟全肽生物科技股份有限公司 董事长曹永圣 合肥远志医药科技开发有限公司 董事长 靳玉明 山东玉满坤生物科技有限公司 董事长
郭 夏 万全医药董事长
宋明选 北京康而福药业有限责任公司 董事长
石党生 浙江福瑞喜药业有限公司 董事长
陈为功 先声药业有限公司 总经理
晋孝通 海南勇毅创新医药科学技术有限公司 总经理
▷14:00-14:15
2024年仿制药行业10大现状—伴随阵阵红包雨王 波 享融智云董事长
▷14:15-16:30
对话决策层 | 企业家/决策层核心观点碰撞
朱海健 力品药业(厦门)股份有限公司 总经理郭大海 诺峰药业/Zennova Pharma董事长兼CEO 蒋 杰 广州国标检验检测有限公司 董事长
葛 磊 西安万凯恩药业有限公司 董事长
姜庆伟 吉林天衡药业有限公司 董事长
唐永红 西安泰科迈科医药技股份有限公司董事长兼总经理
秦 勇 燃点(南京)生物医药科技有限公司 董事长
曹明成 合肥创新医药技术有限公司 董事长
周 俊 天津汇众医药科技发展有限公司 董事长
黄水华 海诚医药集团 董事长、总裁
陶春蕾 安徽万邦医药科技股份有限公司 董事长 吴光美 深圳贝美药业有限公司 董事长/总经理
贺敦伟 则正医药,创始人、董事长兼首席执行官
陈再新 江苏百奥信康医药科技有限公司 董事长
赵 亭 方明药业,总经理
李莉莉 广州科创荣天医药科技有限公司 总经理
焦培想 南京华威医药科技集团有限公司 总经理
周媛烨 广州泓健健康产业有限公司 总经理
魏 鸿 四川宏明博思药业有限公司 总经理
刘 飞 南京迈诺威医药科技有限公司 总经理
8月16日
▷14:30-16:30
对话决策层 | 企业家/决策层核心观点碰撞
陶琇梅 北京诺康达医药科技股份有限公司 创始人,总经理
张 冰 安徽杰玺医药有限公司 董事长
李铁军 山东京卫制药有限公司 董事长
田 元 广州仁恒医药科技股份有限公司 创始人/董事长&总经理
陈西敬 南京菲力康医药科技有限公司 董事长
邓志鸿 山东白云药业有限公司 总经理;山东顺杰健康有限公司董事长
易跃能 湖南易能医药有限公司 董事长
王二敬 天津掌心医药科技有限公司 董事长
高肇林 山东益康药业股份有限公司董事长、总经理
梁晓云 大连美创药业有限公司 总经理
方大兴 华普医药集团,战略BD副总裁
马路军 山西皇城相府药业股份有限公司 董事长兼总经理 王传跃 浙江美华鼎昌医药科技有限公司 董事长
沈一杰 浙江兄弟药业有限公司 总经理
韩冠英 山东丰金生物医药有限公司 副总经理
谭 跃 湖南华纳大药厂股份有限公司(688799) 战略发展总监
立即扫码报名
守正创新中药发展论坛
D馆-D105
▼下滑浏览全部
8月15日
▷9:30-10:00
传承创新,中医药引领健康未来程建明 南京中医药大学药学院教授,江苏省经典名方工程中心主任
▷10:10-10:50
创新中药制剂研发策略探讨
陈大全 烟台大学教授
▷10:50-11:30
医疗机构制剂向创新中药的转化策略柏冬 中国中医科学院中医基础理论研究所中医方证研究中心主任,研究员,博士研究生导师
▷11:30-11:50
中药注射剂再评价难题解析
曹明成 合肥创新医药技术有限公司 董事长
▷14:00-14:40
从中医药理论谈创新中药研发张光霁 浙江中医药大学副书记、二级教授、中医世家
▷14:40-15:20
中药新药物质基础研究和质量标准建立策略
吴佳 上海诗丹德生物技术有限公司 研发部中心主任
▷15:20-16:00
基于丹参三七药对的新药开发姜宝红 中国科学院上海药物研究所研究员
▷16:00-16:40
基于中药有效成分的原创靶点发现寇俊萍 中国药科大学教授
8月16日
▷9:30-10:10
面向产业需求的中药创新研发与转化策略张铁军 天津药物研究院有限公司中药首席专家、和光中药科技(天津)有限公司总经理
▷10:10-10:50
同名同方药临床试验相关问题解析高蕊 中国中医科学院首席研究员、中国中医科学院临床药理研究所副所长
▷10:50-11:30
基于人用经验下的企业新产品开发差异化选品思路探讨尚强 丽珠医药集团 中药研究院常务副院长 四川光大制药有限公司 副总经理
▷11:30-12:10
中药新药的临床价值发现与研发风险管控
姚仲青 珍宝岛药业研发副总
▷14:00-14:30
三结合”审评证据体系下中药新药临床研究樊敏伟 国家中药制药工程技术研究中心 副主任
▷14:30-15:00
大健康需求驱动的药食同源产品研发
辛海量海军军医大学药学系教授/中医系(全国名中医传承工作室)
▷15:00-15:30
中药制剂连续制造一PAT与数学建模技术的研究进展杜若飞 上海中医药大学副研究员
立即扫码报名
改良新药实战案例私享会
D馆-D106
▼下滑浏览全部
8月15日
▷09:30-10:00
改良新药开发策略及案例分析张宁 西交利物浦大学教授,原CDE高级审评员
▷10:00-10:30
药物毛囊靶向递送和新药开发盛晓霞 杭州领业医药科技有限公司总经理
▷10:30-11:00
缓控释微球的工艺与产业化思考
游剑浙江大学教授
▷11:00-11:30
改良型口溶膜的立项与研发案例许建明 浙江和泽医药研究院院长
▷14:00-14:40
一“立”二”改”三“良”,改良型新药上市的三个关键步骤 杨劲 中国药科大学教授
▷14:40-15:20
药械组合产品的开发与思考栾瀚森 和妍(上海)医疗器械有限公司 CTO
▷15:20-16:00
复杂注射剂改良型新药研发关键技术和实例分析
何仲贵沈阳信达泰康医药科技有限公司首席科学家
▷16:00-16:40
儿童改良新药研发案例分享
齐宜广 贝美研发(深圳)有限公司 总经理
8月16日
▷9:30-10:00
透皮制剂改良案例分析—关键质量属性(CQAs)的若干关联性实验与设计
汪晴 大连理工大学教授
▷10:00-10:30
膜剂的改良与创新开发策略陈芳 医药先进制造国家工程研究中心研究员
▷10:30-11:00
改良型新药研发及产业化难点案例分析
吴四清 湖南九维生物医药有限公司创始人&董事长
▷11:00-11:30
难溶性药物口服制剂关键技术及案例分享王泽人 深圳药欣生物联合创始人、董事长和首席科学家
▷11:30-11:50
改良创新药临床试验设计思路和方法 张敏 北京知和爱康医药科技有限公司技术副总经理
▷14:00-14:30
以阿尔兹海默症为例的纳米药物递送研发案例分享高会乐 四川大学教授
▷14:30-15:00
以干眼症为例的眼用制剂研发案例分享与思考张宇 沈阳药科大学教授
▷15:00-15:30
冻干技术在复杂制剂中的应用及案例
刘恒利 凯信远达医药有限公司研发中心负责人
立即扫码报名
仿制药和改良新药立项“稳准狠”论坛
D馆-D107
▼下滑浏览全部
8月15日
▷9:30-10:10
从仿制走向改良:在细分领域挖掘机会王立峰 慧药咨询创始人
▷10:10-10:50
缓释制剂的改良立项及临床开发策略夏燕 百花村医药集团轮值总经理,礼华生物董事长
▷10:50-11:30
基于临床需求的改良型新药立项策略与实施案例吴涛 北京汇诚瑞祥医药生物科技有限公司 首席科学家
▷11:30-12:10
成功上市的505(b)(2)案例对改良新药立项启示陈洪 外专局特聘专家
▷14:00-14:40
以透皮给药技术平台为导向的产品立项及案例分享孙亚洲 长沙晶易医药科技股份有限公司联合创始人、董事、高级工程师、NMPA高级研修学院特聘讲师
▷14:40-15:20
以渗透泵技术平台为导向的产品立项及案例分享石劲敏 汉都医药副总经理
▷15:20-16:00
以眼部给药技术平台为导向的产品立项及案例分享魏继业 苏州惟佑基因生物科技有限公司 董事长
▷16:00-16:40
产业视角下的儿科用药立项策略
赵刚 健民集团研究院 院长
8月16日
▷9:30-10:20
独一无二——以特色、差异化布局选品思路
郭新峰 南京循证生物科技有限公司创始人
▷10:20-11:30
成本优势——以集采品种选品思路张廷杰 风云药谈 创始人
▷11:30-12:10
技术领先——高技术壁垒改良型新药立项及成功案例分享
魏世峰 北京罗诺强施医药技术研发中心有限公司董事长兼CEO
▷13:30-14:30
产品为王——高度内卷之下,仿制药的立项思路与产品布局
魏利军 北京药眼信息咨询有限公司创始人兼CEO
▷14:30-15:00
热火朝天——在中药赛道“稳准狠”立项
王海林 南京恒道医药销售总监,药筛创始人,医药行业数据及政策研究者,药品营销探索者
立即扫码报名
肠外营养复杂制剂项目CDMO论坛
主办单位:盈科海鲲&药融圈
D馆-A01 8月15日 14:30-16:20
▼下滑浏览全部
8月15日
▷14:30-14:55
盈科生物复杂制剂战略思考及产品布局
王利春 研究院院长-副总裁高级工程师,原国家大容量注射剂工程技术研究中心主任,大容量注射剂国家地方联合工程实验室主任
▷14:55-15:05
抽奖
▷15:05-15:30
脂肪乳注射液药学研发要点分享
杨亚妮 博士,医药先进制造国家工程研究中心副研究员,上海市药学会药剂学专委会工业药剂学组组员,上海市药品申评核查专家
▷15:30-15:40
抽奖
▷15:40-16:05
脂肪乳注射剂产业化研究分享
沙琦 盈科生物执行总裁 制药工程硕士,高级工程师,产业化全面管理(泰州&扬州)
立即扫码报名
QCS-CHINA中国药物研发与质量控制大会
主办单位:振强生物&药融圈
D馆-E03 8月15日 10:00-11:40,14:00-16:30
▼下滑浏览全部
8月15日上午
▷ 10:00-10:15
QCS-CHINA主办方:杂质蓝皮书发布
▷10:15-10:20
现场抽奖:杂质蓝皮书赠送
▷10:20-11:00
化药注册申报药学发补常见问题及案例分享
▷11:00-11:40
药物杂质谱分析
胡昌勤 中国食品药品检定研究院原抗生素室主任、化学药品检定首席专家
8月15日下午
▷14:00-14:30
基于审评发补案例分享讨论杂质研究策略周立春 原北京市药品检验鉴定研究所所长助理/教授
▷14:30-15:00
医药杂质标准品稳定性研究
李洋 深圳市江川医药技术有限公司QCS首席科学家
▷15:00-15:10
趣味问答/现场抽奖
▷15:10-15:40
基因毒杂质的风险评估及控制
郑枫 中国药科大学 教授
▷15:40-16:30
【圆桌论坛】
立即扫码报名
国际化原料制剂CDMO论坛
主办单位:汇宇制药&药融圈D馆-D02 8月15日
▼下滑浏览全部
8月15日下午
▷ 14:00-14:10
汇宇专属礼品赠送
▷14:10-14:30
原料药产业化的挑战与应对策略
陈伟东 四川汇宇制药原料药生产总监
▷14:30-14:35
问答/抽奖
▷14:35-14:55
高端复杂制剂研发要点及案例分享
徐上虎 复旦大学药物化学博士,四川汇宇制药研究院二室主任
▷14:55-15:00
有奖问答/抽奖
▷15:00-15:20
复杂注射剂的产业化关键与难点
任永春 四川汇宇制药副总裁兼生产基地总经理
▷15:20-15:25
有奖问答/抽奖
▷14:30-14:45
原料药中美双报差异
江 霞 灵生益国际注册总监
▷14:30-14:45
国际化出海平台介绍
▷15:45-15:55
圆桌论坛:高端制剂如何破局-进入出海热潮?
中美欧研发、生产、注册、审评审批之路
立即扫码报名
CDMO产业创新合作大会
主办单位:华益药业&药融圈
D馆-E01 8月15日下午
▼下滑浏览全部
8月15日下午
▷ 13:40-14:05
MAH制度下受托生产企业遴选和审核实践
黄小枫
华益药业质量受权人
▷14:05-14:10
有奖问答
▷14:10-14:30
受托生产企业的遴选-技术转移实战经验分享
操铖
华益药业研发总监
▷14:05-14:10
趣味游戏
▷14:10-14:30
优质项目推介
史薇
益药业项目调研总监
▷14:30-14:45
新产品引入后的工艺验证和清洁验证
孙结华
华益药业质量负责人
▷14:30-14:45
药品共线生产风险评估一高活性产品评估
李玉宝
华益药业质保中心总监
▷14:30-14:45
有奖问答
立即扫码报名
CRO创新模式,助力高效研发
主办单位:万邦医药&药融圈
D馆-C03 8月15-16日
▼下滑浏览全部
8月15日
▷ 09:00-9:30
外用皮质类固醇激素类药物的PD-BE试验
余玲玲
临床研究中心项目负责人
▷10:00-10:30
维生素类药物的液相色谱质谱生物分析
许杨
安徽万邦副总经理
▷14:00-14:30
安徽万邦自研项目推介
陈斌
安徽万邦技术总监/沈阳药科大学博士
▷15:00-15:30
食品类人体试食试验交流讨论
邵凤
安徽万邦副总经理
8月16日
▷09:30-10:00
体外诊断试剂临床试验实施要点
顾晓恒
安徽万邦医疗器械临床评价负责人
▷10:30-11:00
妇科领域验证性临床项目交流讨论
朱舟沁
安徽万邦运营总监
立即扫码报名
加速创新-药物研发与转化论坛
主办单位:都正生物&药融圈
D馆-E05-1 8月15日
▼下滑浏览全部
▷ 14:00-14:30
【线上】3类仿制药立项风险与临床研究策略选择
欧阳冬生 长沙都正生物科技股份有限公司董事长
▷14:30-14:35
抽奖
▷14:35-14:50
临床试验一站式解决方案
钟珺 长沙都正生物科技股份有限公司商务发展中心副总监
▷14:50-15:10
改良型新药立项研发的挑战
王欣桐
长沙都正生物科技股份有限公司医学事务中心总监
▷15:10-15:15
抽奖
▷15:15-15:40
模型引导的创新药给药策略制定
巨舸航 长沙砝码柯数据科技有限责任公司,定量药理分析师
▷15:40-15:45
抽奖
▷15:45-16:10
【线上】智慧实验室平台:药物研发领域的智能加速器”
张 均 长沙通诺信息科技有限责任公司售前经理
立即扫码报名
E馆-制药产业链馆
原料药企业家年会
E馆-E105 8月15日-8月16日
▼下滑浏览全部
8月15日主持人:彭利增,济南爱思创始人
▷13:30-13:35
李湘 中肽生化,创始人兼董事长
▷13:35-13:40
陈恬 车头制药,董事长
▷13:40-13:45
叶伟平 华先医药/莱佛士制药 董事长
▷13:45-13:50
黄勇刚 汉泰生物,董事长
▷13:50-13:55
赵磊 四川青木制药,总经理
▷14:00-14:05
魏彦君 威智医药,董事长
▷14:05-14:10
邹 平 海门慧聚,总经理
▷14:10-14:15
燕立波 开元医药,CEO
▷14:15-14:20
叶华华 杭州维华生物,董事长
▷14:20-14:25
刘建 瑞德林生物,联合创始人兼CEO
▷14:25-14:30
张鑫磊 北京屹柏生物
▷14:30-14:35
宋文志 上海棓诺,总经理
▷14:35-14:40
张小顺 安徽海康药业,董事长
▷14:40-14:45
茅仲平 汉德创宏,董事长
▷14:45-14:50
梁承 广西汉和药业,董事长
▷14:50-14:55
马士忠 瀚鸿科技,总经理
▷14:55-15:00
徐俊 正济药业,董事长
▷15:00-15:05
孙建华 南京贝杰,董事长
▷15:05-15:10
陈正杰 山东微研生物,总经理
▷15:10-15:15
薛吉军 皓天制药,董事长
▷15:15-15:20
杨玉超 南京安易科化工,董事长
▷15:20-15:25
嘉宾行程确定中...
▷15:25-15:30
杨勇泰德制药 副总裁
▷15:30-15:35
袁永坤 苏州亚科,董事长
▷15:35-15:40
王征野 苏州传健医药 董事长
8月16日
▷09:00-09:05
张颢腾 基诺厚普生物,执行长
▷09:05-09:10
金春梅 大连奥川生物,董事长
▷09:10-09:15
王雷 都创医药,CEO
▷09:15-09:20
赵志仁 安徽优雅化工&盐城格瑞茵化工,总经理
▷09:20-09:25
范勇 瑞宝恩科技,董事长
▷09:25-09:30
范志伟 宁夏金维,总经理
▷09:30-09:35
吴彦 微远生物,董事长
▷09:35-09:40
李明华 济坤生物,董事长
▷09:40-09:45
李一彪 东岸生物,董事长
▷09:45-09:50
蔡凡平 阜新孚隆宝医药,总经理
▷09:50-09:55
傅鹏 木槿化学CEO,核心创始人
▷10:00-10:05
张林宝 坤宝股份,董事长
▷10:05-10:10
何建国南松凯博生物制药,董事长
▷10:10-10:15
董来山安徽峆一药业股份有限公司,董事长
▷10:15-10:20
罗前东成都普康生物,董事长
▷10:20-10:25
李永南方制药,总经理
▷10:25-10:30
王向旭杭州铭辰医药,总经理
▷10:30-10:35
郭书明开封明仁,董事长
▷10:35-10:40
蔡伟兵浦拉司科技,创始人
▷10:40-10:45
乔木云合成,总经理
▷10:45-10:50
陈成苏州纳美特
▷10:50-10:55
梁永宏上海相辉医药 联合创始人兼首席执行官
▷11:00-11:05
杨晓柯开封达瑞药业 董事长
▷11:05-11:10
嘉宾行程确定中...
▷11:10-11:15
姜能桥武汉康蓝药业
▷11:15-11:20
崔家荣 萃英化学 上海研发中心负责人
▷11:2-11:25
陈维华 上海力田化学 董事长
立即扫码报名
绿色制药技术大会
E馆-E103 8月15-16日
▼下滑浏览全部
8月15日主持人:王鹏 杭州国瑞生物 总经理
▷09:20-09:40
高端化学品制造的微反应技术一一过程强化和安全,mRNA疫苗高效递送系统陈光文 中国科学院大连化学物理研究所
▷09:40-10:00
高效催化促进药物绿色制造汤文军中国科学院上海有机化学研究所研究员、国科大杭州高等研究院首席教授
▷10:00-10:15
微生物合成生物技术驱动原料药产业重塑
李永泉 浙大求是特聘教授,浙大微生物学系主任/浙大药物生物技术研究所所长
▷10:15-10:30
连续流在药物研究中的应用曹国斌菲立化学 常务副总
▷10:30-10:50
算法驱动的生物系统优化晁然衍进科技 联合创始人兼CEO
▷10:50-11:10
光化学连续流技术在药物开发中的应用张亮 普渡机械 研发负责人
▷11:10-11:30
生物技术在小分子药物合成中的应用
马建国 维亚生物高级副总裁兼朗华制药首席执行官
▷11:45-13:30
午餐
▷13:30-13:45
化学工艺降本策略:短路线、高产率、联产设计陈从喜北京颖诺凯胜科技/分子树 创始人CEO兼CSO
▷13:45-14:00
维生素D系列药物的开发李毅甘肃皓天科技 研究院院长
▷14:00-14:20
不对称催化氢化、环氧化技术开发及应用
钟为慧 浙江工业大学药学院 教授、博导,药物合成工艺研究所 所长
▷14:20-14:35
微通道反应器在高端化工领域的应用及特点
李钰龙 山东特创新材料科技有限公司 创始人兼总经理
▷14:35-14:50
对微化工技术及应用的一点体会金英泽 欧世盛科技 董事长兼CEO
▷14:50-15:10
生物学平台-在医药大健康领域中的应用王筱欣贝莱生物 CEO
▷15:10-15:25
透彻杂质谱研究对简化GLP-1侧链生产工艺的帮助刘国柱 长沙晨辰医药 董事长、创始人
▷15:25-15:40
内卷时代对工艺开发及优化的一些思考葛敏 江苏正济药业 首席科学家、副总裁
▷15:40-16:00
提取加半合成原料药工艺案例分享彭学东 威胜生物医药 联合创始人
▷16:00-16:15
医药结晶中的杂质、晶型、制剂等问题探究龚俊波 天津大学国家工业结晶工程技术研究中心主任,教授
8月16日主持人:王鹏 杭州国瑞生物,总经理
▷09:30-09:45
如何设计打造安全、环保的API多功能生产线
刘万斌 浙江六合 董事长
▷09:45-10:00
过渡金属催化反应在手性药物制备中的应用
赵卉欣诺科催化剂
▷10:00-10:15
光化学产业化
李一鸣 善施科技 董事长
▷10:15-10:30
微填充床反应器连续加氢技术在精细化学品合成中的应用
陈兴坤 相流(北京)科技有限公司 技术顾问
▷10:30-10:45
新药IND阶段原料药工艺研发的关注点和降本增效策略(case分享)
邱小龙 美迪西 工艺部执行主任
▷10:45-11:00
连续流技术助力绿色化学自动化
李鹏飞 石榴化工科技 总经理
▷11:00-11:15
微波法第二代分子筛膜在制药行业溶媒回用和高纯化学品提纯精制中的应用
张立峰 浙江汇甬新材料有限公司 总经理
▷11:15-11:40
实现化学工艺过程中最有价值的连续
马兵安微(惠和化德)
立即扫码报名
IPO掌门人说
E馆-E105 8月15日上午
议程更新中,敬请期待...
立即扫码报名
国际原料药供应链对接会
E馆-E102 8月15日
▼下滑浏览全部
主持人:王秋月 GSK,国际采购
▷09:30-10:00
秦为民 STADA中国区采购负责人
▷09:40-09:50
刘先生 美国Biotech,国内采购负责人
▷09:50-10:00
黄勇刚 汉泰生物,董事长
▷10:00-10:10
史宏伟 国药国际
▷10:10-10:20
王秋月 GSK,国际采购
▷10:20-10:30
余小兵 广州安信医药,创始人
▷10:30-10:40
汤敏钟 维振纬嘉,总经理
▷10:40-10:50
谈建伟 龙圣药业,采购负责人
▷11:00-11:40
何君伟 美印医药科技 总经理(主持人)
胡顺良 浙江省医药保健品进出口有限责任公司 副总经理
赵志仁 安徽优雅化工&盐城格瑞茵化工 总经理
方贤威 浙江睿盈维生物 总经理
燕立波 开元医药 CEO
▷13:30-13:40
齐琼sourcing manager/SCM Dept.采购经理/供应链管理部
▷13:40-13:50
周剑侠Hikma,Country Manager-China
▷13:50-14:00
李丽芳石家庄鼎旻医药 董事长
▷14:00-14:10
金炜Group Procurement Raw Materials Souring Head,China Euroapi
▷14:10-14:20
唐威宇 雅培战略采购负责人
▷14:20-14:30
秦玲丽 axplora,采购经理
▷14:30-14:40
杨勇 Medichem,Head of China
▷14:40-14:50
王云 Midas Pharma,中国总经理
▷14:50-15:00
邹琳 MOEHS,采购经理
▷15:00-15:10
袁先生资深行业嘉宾
▷15:10-15:20
许兴明Zentiva 采购顾问
▷15:20-16:00
秦为民 STADA中国区采购,负责人(主持)
谭满芳 明月药业/明药生物,创始人
金春梅 大连奥川生物,董事长
陈智勇 日本Chori蝶理商社,医药负责人
叶 婷 杭州拜邦生物
立即扫码报名
口服固体缓控释制剂大会
E馆-E101 8月15-16日
▼下滑浏览全部
8月15日
▷9:30-10:00
基于临床需求的口服缓控释制剂研发高申 上海长海医院,主任药师
▷10:00-10:30
口服多肽药物新型递送系统的研究史亚楠烟台大学教授
▷10:30-11:00
连续制造视角下的双螺杆热熔挤出和连续制粒杨高品苏州璞佩珊科技有限公司总经理
▷11:00-11:20
缓控释制剂开发重难点剖析及案例分享施斌则正医药CTO
▷11:20-11:40
制剂中药物晶型和粒度分析的创新方法
谢治平 福州华为医药技术开发有限公司高级研究员
▷14:00-14:20
复方口服固体制剂技术要点李明丽济宁学院教授 山东诺明康药物研究院有限公司创始人
▷14:20-14:50
固体分散体设计及开发的基础应用研究蔡挺中国药科大学教授
▷14:50-15:10
制粒、包衣技术创新研究与应用韩盈龙宜春万申智能装备股份有限公司 固体制剂事业部技术经理
▷15:10-15:40
缓控释制剂的技术可行性评估和处方工艺的选择李想 广州玻思韬控释药业有限公司制剂执行总监
▷15:40-16:00
用于缓释口服固体制剂的全配方创新辅料杨继荣卡乐康中国释药配方技术经理
8月16日
▷09:30-10:00
口服缓控释制剂:从理论到实践付强沈阳药科大学教授
▷10:00-10:30
仿制药保质量顾成本案例分享--质量和成本源于设计雷继峰上海安必生制药技术有限公司董事长
▷10:30-11:00
口服缓控释制剂的一致性评价重点和策略探讨梁超峰广东省药学会药用辅料专委会副主任委员主任药师
▷11:00-11:30
缓释制剂溶出性质模型化研究—以盐酸青藤碱缓释制剂为李文龙天津中医药大学研究员
立即扫码报名
中国药用辅料大会
E馆-E102 8月16日
▼下滑浏览全部
▷09:30-10:10
药品上市后药用辅料变更研究的技术考量刘雁鸣湖南省药品检验检测研究院副院长
▷10:10-10:50
纳米制剂关键辅料与质控策略孙春萌中国药科大学教授
▷10:50-11:30
药用辅料登记及辅料关联审评审批李银博悉咨医药联合创办人
▷11:30-11:50
强阻隔薄膜包衣预混剂的研究和应用杨学成上海新菲尔生物制药工程技术有限公司,副总经理
▷14:00-14:40
药用辅料乳糖质量研究进展及应用前景袁耀佐 江苏省食品药品监督检验研究院,检验技术研究室主任
▷14:40-15:20
以口服固体制剂为例的关键药用辅料应用陈英广东省药品检验所辅料室负责人
立即扫码报名
医药全渠道营销与新药商业化落地
E馆-E106 8月15-16日
▼下滑浏览全部
8月15日上午医药全渠道营销
▷9:30-10:00
后MAH时代中国医药市场发展趋势分析陈一进麦池书院理事长MAH专家
▷10:00-10:45
医改背后的政治经济学逻辑张 瑞 杭州六倍体科技(药脉通趣学术)董事长
▷10:45-11:30
集采反腐之下,院内处方药推广迭代之路王鹏.王P药脉通趣学术副总,《医药代表专业化指南》作者
▷11:30-12:00
线上线下融合,中国医药零售大变局金林志豫医药人俱乐部会长
8月15日下午医药产品的商业化落地策略
▷14:00-14:45
中成药学术体系搭建与合规推广赵佳震远大集团多普泰药业市场总监,《处方药合规推广实战宝典》作者
▷14:45-15:30
模式之战——决胜第三终端市场张江民第三终端产品操盘手,诊所动销畅销书作者
▷15:30-16:15
《以患者为中心的全渠道商策略落地》袁 江 求恩医药创始人
▷16:15-17:00
专业的区域CS0如何帮助产品快速发展?陈 新广州新健元医药总经理
8月16日上午创新药的高效专业化推广
▷9:30-10:30
创新药企,如何打造可控高发展的学术化推广体系-区域学术队伍打造与新工具的使用王鹏.王P药脉通趣学术副总,《医药代表专业化指南》作者
▷10:30-11:15
新时期阳光化准入的新打法孙坚彤院企双跨专家京省级三甲医院药剂科主任
▷11:15-12:00
院外非医保产品市场开拓张冬梅上海凤栖桐生物科技有限公司董事长
8月16日下午批文交易大会
立即扫码报名
提升原料药中间体企业新质生产力——开拓行业新天地主办单位:新天地&药融圈
E馆-E06 8月15日
▼下滑浏览全部
圆桌:原料药企业的创新转型之路-合成生物学
1.酶催化、合成生物技术如何赋能传统企业
2.传统企业布局合成案例或产品
3.传统企业如何与生物学企业结合实现降本增效,找到新的增长点
嘉宾阵容
刘超 新天地药业总工程师
竺伟 尚科生物 CEO/首席科学家
倪国伟 上海微理智成生物 创始人
焦银山 杭州合碳创物 创始人
罗志波 杭州微远生物 联合创始人兼CTO
圆桌:创新·合作·共赢:探索制药产业链一体化的未来”
1.国内制药产业链内卷or出海抉择
2.国际制药产业链发展现状及供应链"去中国化!
3.中间体原料药制剂一体化发展的机会与挑战
嘉宾阵容
刘超 新天地药业总工程师
吕新河上海彼特化工 董事长
秦为民 STADA中国区 采购负责人
蒋兴凯 睿铼医药 董事长
魏博 则正医药 首席商务官
立即扫码报名
“不可思议”搭档——原料药VS合成生物学主办单位:荣耀生物&药融圈
E馆-B02 8月15日 10:30-16:00
▼下滑浏览全部
话题:
1.原料药中间体企业与合成生物学结合优势?
2.原料药企业如何切入合成生物学及案例分享?
3.原料药及合成生物学企业如何立项?
拟邀嘉宾
陈仁尔荣耀生物 创始人
王 勇诺云生物 创始人
吴 彦微远生物 创始人
肖延铭英沃迪生物 创始人
徐应木佰依源生物 创始人
立即扫码报名
原料药中间体降本增效——”酶“你不行
主办单位:尚科生物&药融圈E馆-C02 8月15日 14:00
▼下滑浏览全部
嘉宾阵容
竺 伟(主持人)尚科生物医药(上海)有限公司 总经理
程志刚湖北亨迪药业股份有限公司 董事长
郭宏明江苏惠利生物科技有限公司 董事长
柯 威南京工业大学产业教授、南京膜材料产业技术研究员 研发总监
李 刚山东四环药业股份有限公司 董事长
立即扫码报名
开拓创新,合成未来
主办单位:拓新药业&药融圈E-D07 8月15日 13:30-14:15
嘉宾阵容
王文革 江太生物 创始任(主持人)
吕洪志河北齐达生物科技有限公司 总经理
张宝琪百开盛(上海)生物科技有限公司 总助
罗 玮江南大学教授
立即扫码报名
IBS圆桌会:寻找增量的原力
联合主办:Flamma S.P.a.富乐马集团,汇宇制药,增量原力咨询8月14日
▼下滑浏览全部
▷17:00-17:30
嘉宾签到
▷17:30-17:35
主办方致辞
▷17:35-17:40
联合主办致辞
▷17:40-17:50
中国有没有机会?反向出海,从意大利到中国
▷17:50-18:00
出海真的容易吗?中国药企征战欧洲市场的经验
▷18:00-18:10
CRO怎么活下去,从Biotech到CRO,扭亏为盈的尝试
▷18:10-18:20
融资过冬?现在这行情下,哪些项目还能心动掏钱?
▷18:20-18:30
裁员还是替代?ChatGPT时代,你的公司究竟还需要多少员工?
▷18:30-13:55
用餐及社交
立即扫码报名
医药化工企业家分享会
活动主办:普渡生物苏州凯悦酒店 8月14日
▷15:00-16:00
签到
▷16:00-16:15
分享会欢迎致辞
▷16:15-17:30
企业家观点分享&供需发布
▷17:30-18:00
自由交流
▷18:00-20:00
合作交流晚宴
立即扫码报名
跟着我们云逛展
!
想参与CMC-China博览会又没时间亲临现场的你,主办方邀你在线“云”看展!本次云看展开将会有专业主持人带领大家走进仅三生物、百事兴、四川汇宇、壹生科、新济药业等等企业,探索展会第一现场,并且部分展位还设置了精彩的展位活动,邀请了行业大咖们进行专题分享和圆桌讨论,,届时参与直播互动还有机会抽取精美礼品!点击立即预约!
❖点击下列图片,即可电话预定,报“CMC-China博览会”即可享受住宿协议价❖以下根据网络信息推荐附近酒店,信息仅供参考,请参会嘉宾根据需要自行联系酒店或通过“携程”等预订:
价格区间:700+元
价格区间:500-700元
价格区间:400-500元
价格区间:200-400元
展商名录向上滑动阅览
参会企业名录 向上滑动阅览
END
苏州国际博览中心
2024年8月15-16日
抗体药物偶联物申请上市临床结果
100 项与 星尘生物科技(上海)有限公司 相关的药物交易
登录后查看更多信息
100 项与 星尘生物科技(上海)有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年11月06日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
药物发现
2
8
临床前
临床申请
3
1
临床申请批准
临床1期
3
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
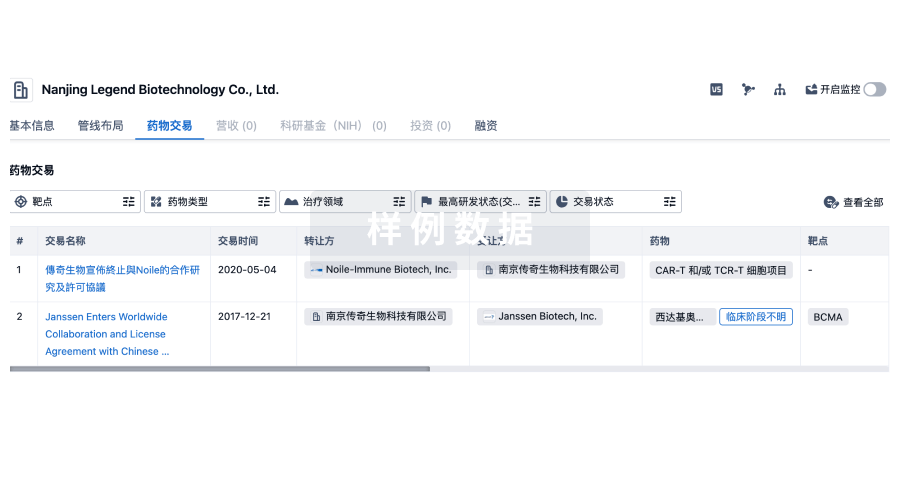
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
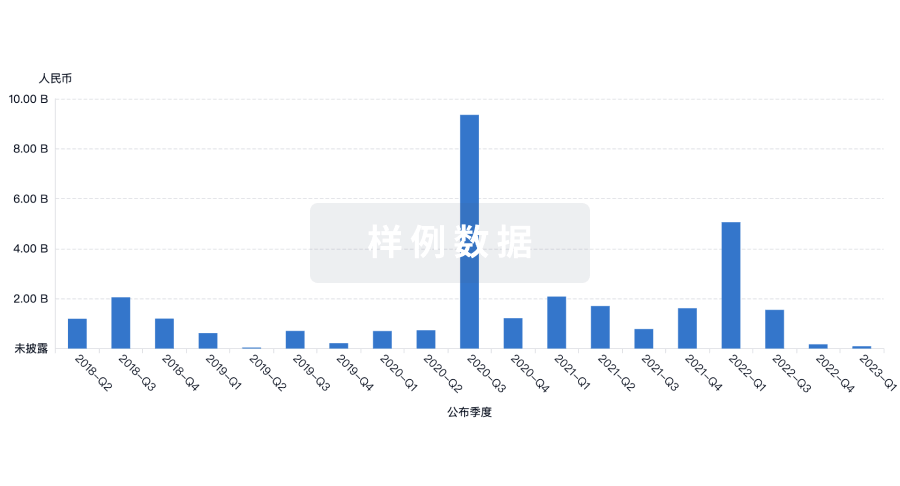
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用