预约演示
更新于:2025-05-07

Scopus Biopharma, Inc.
更新于:2025-05-07
概览
标签
肿瘤
血液及淋巴系统疾病
泌尿生殖系统疾病
ASO
CpG ODN
siRNA
疾病领域得分
一眼洞穿机构专注的疾病领域

暂无数据
技术平台
公司药物应用最多的技术
暂无数据
靶点
公司最常开发的靶点
暂无数据
| 排名前五的药物类型 | 数量 |
|---|---|
| ASO | 4 |
| CpG ODN | 3 |
| 小分子化药 | 1 |
| siRNA | 1 |
关联
9
项与 Scopus Biopharma, Inc. 相关的药物靶点 |
作用机制 STAT3抑制剂 |
非在研适应症- |
最高研发阶段临床前 |
首次获批国家/地区- |
首次获批日期- |
靶点 |
作用机制 WDR5 抑制剂 |
在研适应症 |
非在研适应症- |
最高研发阶段临床前 |
首次获批国家/地区- |
首次获批日期- |


作用机制 STAT3抑制剂 [+1] |
在研适应症 |
非在研适应症- |
最高研发阶段临床前 |
首次获批国家/地区- |
首次获批日期- |
100 项与 Scopus Biopharma, Inc. 相关的临床结果
登录后查看更多信息
0 项与 Scopus Biopharma, Inc. 相关的专利(医药)
登录后查看更多信息
25
项与 Scopus Biopharma, Inc. 相关的新闻(医药)2025-02-12
·梅斯医学
尿道下裂(hypospadias)是一种常见的阴茎畸形,男性尿道开口位置异常的先天畸形,尿道口未开口在阴茎龟头的顶端,而是开口在阴茎腹侧正常尿道口至会阴部的不同部位,部分病例伴发阴茎下弯。尿道下裂是一个先天性的疾病,不会随着患儿年龄的增长而自行好转。
据数据显示,在活产男婴中的尿道下裂发病率为0.3%~0.7%,即每300个男孩子就有1个是尿道下裂,而且这个概率还在不断上升。虽然尿道下裂的发病率不高,但关于超声在产前诊断尿道下裂的作用和价值,观点多样。尽管尿道下裂并非致命性畸形,但较高程度的严重性对儿童的生理和心理都有显著影响。
近期,来自我国重庆医科大学附属儿童医院泌尿外科研究人员在eClinicalMedicine杂志上发表了题为“Performance of ultrasound in detecting fetal hypospadias during pregnancy: a pooled analysis”的一项研究,旨在评估超声在尿道下裂产前诊断中的价值。
在这项系统评价和荟萃分析中,研究人员检索了PubMed、Web of Science、Scopus、Embase以及中国知网(CNKI)数据库直至2024年6月30日。关键词包括:尿道下裂、超声、产前诊断。根据纳入标准对研究进行评估,并通过标准化方案提取纳入的研究数据。主要结果集中在超声检测胎儿尿道下裂的阳性预测值(PPV)、准确性、敏感性和特异性。使用诊断准确性研究质量评估工具-2(QUADAS-2)量表评估偏倚风险。采用Bootstrap方法计算PPV。当合并的研究少于或等于五项时,采用基于 Hartung-Knapp-Sidik-Jonkman (HKSJ) 方法的随机效应模型作为荟萃分析策略来评估总体效应。敏感性和特异性通过综合受试者操作特征(SROC)曲线进行汇总。
结果显示,系统性回顾共纳入了 9 项研究,600 个病例。纳入研究的孕周中位数为(27.64 ± 3.15)周。二维超声(2DUS)在诊断胎儿尿道下裂和外生殖器畸形的总体PPV分别为81%(95%置信区间(CI):77%-85%)和88%(95% CI:86%-91%)。
对五项研究深入分析后发现,2DUS联合三维超声(3DUS)的准确性为84%(95% CI:78%-89%),而单独使用2DUS的准确性为74%(95% CI:69%-78%),两者之间差异为10%。2DUS结合3DUS诊断胎儿尿道下裂的合并敏感性为86%(95% CI:79%-93%),特异性为77%(95% CI:69%-86%),曲线下面积(AUC)为0.86(95% CI:0.83-0.89)。
总之,这是首次全面评估妊娠期超声检测胎儿尿道下裂的诊断荟萃分析,表明超声在产前诊断尿道下裂方面确实具有重要价值。未来需要开展更多的相关研究来验证现有研究结果,并为提升临床实践水平提供更为详尽和系统的指导建议。
原始出处
Zhang, Qiang et al.Performance of ultrasound in detecting fetal hypospadias during pregnancy: a pooled analysis.eClinicalMedicine, Volume 81, 103091
撰文 | 梅斯医学
编辑 | 阿拉斯加宝
● 茶+咖啡更续命!我国学者研究:每天喝3杯茶,抗衰效果最好;茶和咖啡同饮,全因死亡风险降低22%,骨质疏松风险降低32%
●每周吃几个鸡蛋合适?多项研究:每周1-6个,心血管死亡风险降低29%,全因死亡风险降低15%;但超过6个,全因死亡风险增加35%
● 热议!医院行政人员首次发声:总说临床养活行政后勤,我们为了医院也耗尽心力!三明模式下,行政后勤不像以前吃香了,到手工资大降
版权说明:梅斯医学(MedSci)是国内领先的医学科研与学术服务平台,致力于医疗质量的改进,为临床实践提供智慧、精准的决策支持,让医生与患者受益。欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。
点击下方「阅读原文」 立刻下载梅斯医学APP!
2024-12-20
·智药邦
论文概览
人工智能时代下的蛋白质从头设计
刘南1,金小程1,杨崇周2,王梓洋2,闵小平2*,葛胜祥1*
1 厦门大学 公共卫生学院 分子疫苗学与分子诊断国家重点实验室
2 厦门大学 信息学院 人工智能研究院
摘要:具有特定功能和特性的蛋白质在生物医药、纳米材料等领域至关重要。蛋白质从头设计能够定制序列以生成具有所需结构、自然界中未存在的蛋白质。近年来,随着人工智能的迅猛发展,深度学习生成模型逐渐成为强大工具,许多功能性蛋白质的设计都达到了原子级别的精度。本文概述了蛋白质从头设计的演进,着重介绍了其最新算法模型,并分析了其存在的问题,如设计成功率低、精度不足以及对实验验证的依赖性,最后探讨了蛋白质设计的未来趋势,旨在为研究者和从业者提供有益参考。
在地球生命演化的40亿年中形成了功能丰富的蛋白质,这些蛋白质作为功能性分子,在DNA/RNA复制、转录、翻译等生命活动中起着至关重要的作用,是执行生命功能的核心元素[1]。根据热力学原理[2],蛋白质的功能由结构决定,而其三维结构由20种氨基酸序列排列所确定。然而,蛋白质序列多样性极高,一个由200个氨基酸组成的蛋白质能有20200种可能[3],超过宇宙原子总和[4]。自然演化形成了其中的一部分序列,组成了众多的蛋白家族,但利用传统方法如随机突变设计蛋白质面临巨大挑战[3]。近年来,蛋白质从头设计取得了显著进展,可以不依赖天然模板设计新型蛋白质,这为探索具有特定功能的全新蛋白质、满足人类不断增长的需求打开了大门。
蛋白质从头设计一般包括两个步骤:蛋白质主链三级结构的生成,残基位置及侧链构象设计[5]。这两步的基础是能量函数的评估。1980年,研究人员提出了蛋白质折叠的最低能量状态假说,认为氨基酸序列决定了其三维结构[2]。因此,传统设计方法主要依据能量最低原理来寻找合适的能量函数。典型的研究方法有RosettaDesign[6]、CHARMM[7]、ISAMBARD[8]、FoldX[9]等。Rosetta提供了最常用的一种能量函数,涵盖了物理的能量组成和确定蛋白质结构的实验因素,包括氢键网络[10]、范德华力[11]、极性及疏水性[12]等,并采用蒙特卡罗模拟退火、死码消除算法、遗传算法和优化理论等[13-16]方法来优化设计结果。在过去的40年中,基于传统方法的蛋白质从头设计成功率低且依赖于多轮实验验证以确保其可靠性。
近10年来,随着计算能力的提升、蛋白质数据的累积以及人工智能(artificial intelligence, AI)技术的应用,蛋白质从头设计发生了根本性的变革。设计方法由基于第一性原理的传统方式转变成了运用深度学习的现代方法,特别是AlphaFold2[17]及RoseTTAFold[18]在蛋白质结构预测上的开创性成就。据报道,通过整合最新的计算设计、筛选方法,实验室工作量可大幅减少至万分之一[19]。2023年12月,David Baker展示了新型扩散模型RFdiffusion,其在Bim和PTH体系的应用表明所设计的多肽结合蛋白具有高度亲和力,达到pmol量级[20]。虽有成功实例,但蛋白质从头设计依旧面临挑战,包括低成功率、计算模型局限性以及对实验验证的依赖等。另外,未来的研究中需关注的问题还包括缩短设计周期和降低成本。
总体而言,蛋白质从头设计在先进算法的助力下仍具有巨大的发展潜力,该技术在癌症治疗、纳米技术、下一代人工智能疫苗、生物兼容性材料等众多领域均显示出巨大潜能(图1)。本文回顾蛋白质从头设计的发展历程和最新成就,分析现存挑战,探讨未来的发展方向,旨在为该领域的研究人员提供参考。
图1 蛋白质从头设计的应用
>>>>
1 蛋白质从头设计概述
1.1 蛋白质从头设计演变
蛋白质从头设计涉及的序列以及构象空间极为复杂,曾被视为不可能完成的任务[21]。自蛋白质从头设计首次被提出以来[22],该领域面临诸多挑战。蛋白质的从头设计最初是指从头开始设计蛋白质,而不是修改天然存在的蛋白质[23]。随着蛋白质数据库扩充以及计算能力和算法的迭代更新,蛋白质从头设计的概念不断拓展。在该领域的发展历程中,有研究者将其前20多年演变过程分成3个阶段[24]:依赖物理模型设计、以物理化学原理为导向的计算设计以及结合片段和生物信息学的设计,并回顾了该领域的里程碑进展和成就。同时,有研究者提出了最小化蛋白设计、理性蛋白设计、蛋白质计算设计这3种蛋白设计方法[25],并制定了详尽的时间表来描述从头设计肽及蛋白质的进展。上述工作总结了蛋白质从头设计的发展历程,虽然时间线不同,但是各阶段和定义方法基本一致。
近10年间,随着对蛋白质折叠机制理解的不断深入,蛋白质结构预测的准确性显著提高,使得设计的氨基酸序列能够折叠成与晶体结构高度一致的蛋白质构象。特别在2021年,深度学习驱动的AlphaFold2[17]在蛋白质结构预测中实现了突破,彰显了深度学习的巨大优势。同时,蛋白质从头设计领域也产生了包括病毒抑制剂[26-27]、萤光素酶[28]、ProteinMPNN[29]、RFdiffusion[20]等在内的令人振奋的成果及设计工具,开启了新的发展阶段。AlphaFold2的出现标志着蛋白质设计领域的重大转折点,本文据此将蛋白质从头设计策略分为传统和基于AI两大类别(图2)。
图2 1970–2024年的蛋白质从头设计发展史
1.2 传统蛋白质从头设计基本步骤
传统蛋白质从头设计通常利用能量函数和搜索策略揭示序列、结构和功能之间的关系,该过程分为4个阶段[30]:(1) 定义目标蛋白结构;(2) 生成主链骨架;(3) 确定氨基酸序列;(4) 评估设计序列与结构的兼容性(图3)。
图3 蛋白质设计流程图
从头设计从确定目标蛋白拓扑结构入手,考虑诸如二级结构的元素、连接形状和几何形状等因素。接下来,筛选更高度设计性的结构特征,获得优选构型。骨架构建通常基于蛋白质数据库(protein database, PDB)的结构信息,方法包括片段组装[31]、参数化设计[32]、kinematic closure[33]、原生结构图扩展[34]、循环构建[35]等。当前,依靠已有片段组装是最成功的通用方法,例如Rosetta TopoBuilder[36]可以构建各种蛋白质构型,包括微型蛋白[37],尽管其难度会随着主干结构元素增加而升高。
为选定骨架结构寻找能量最小化的氨基酸序列需要进行序列填充设计。大部分序列设计算法依赖于能量函数,如David Baker课题组的Rosetta软件中广泛应用的Packer[38]以及刘海燕课题组开发的ABACUS方法[39-40]。Packer主要用于模拟退火处理主链的侧链放置,以确定最佳序列顺序。研究人员设计了一系列偏向策略[41-42],包含层设计[43]、氨基酸组成限制[44]、二硫键搜索[45]、自定义选择关键结构特征的氨基酸[46]等,以偏向于最佳设计方案,减少能量函数和采样算法的局限性。经过Rosetta的快速属性评估与排名,这些方法能较大概率地产生结构稳定且符合目标的设计。
尽管得到低能序列有利于折叠成目标结构,但还需进一步评估设计序列与目标结构的兼容性以确保正确折叠。AlphaFold2出现前,高端的结构预测技术[36]由于不准确、成本高昂等缺点促使人们选择成本较低的序列结构兼容性测试,通常分为局部和非局部验证,但这种方法仅限于少数设计的验证。
1.3 传统方法的局限性
在过去的几十年里,蛋白质设计领域经历了从依赖经验规则到计算驱动方法的显著转变。这一进步不仅将原本难以捉摸的理念转化为现实,而且对精准医疗[27]、疫苗研发[47-48]、纳米材料[49]领域产生了深远影响。虽然传统从头设计能够解决一些挑战性任务,但是人们逐渐认识到其准确性和稳定性有限。传统从头设计受到能量函数描述不精确及搜索策略局限的影响,存在计算量大、很难找到最优解、成功率低、精度不足、能量函数本身立场精度等问题,通常需要通过实验方法的优化来验证设计的可靠性。此外,这些方法通常仅适用于特定生物系统,难以应用于不同生物学问题的迁移设计。
>>>>
2 蛋白质从头设计算法模型
计算能力的显著提升和高通量技术的应用为蛋白质数据的积累提供了强大支持,推动了蛋白质从头设计从传统方法向数据驱动的计算方法转变[50]。近年来,深度学习技术在计算机视觉、自然语言处理(natural language processing, NLP)等[51]领域的应用已逐步成熟。由于数据结构和处理逻辑的相似性,这些先进技术正对生物科学领域产生深远影响。例如氨基酸序列类似于人类语言,现有的NLP模型便能迅速转化为蛋白质序列的有效参数化工具。
在大量蛋白质序列和结构数据的支持下,深度学习在探索序列-结构-功能关系方面取得了重要进展,革新了功能性蛋白的设计方法。基于深度学习的蛋白质从头设计主要分为两大类:第一类基于骨架结构的蛋白设计,包括固定主链和可变骨架的序列优化[52];第二类不依赖于结构的直接序列生成,用于探索序列空间和蛋白质生成。
2.1 基于骨架结构的蛋白设计
在蛋白质结构预测领域,深度学习从早期的残基接触预测和辅助结构建模[53-57],到准确预测残基的几何性质和基于几何约束的蛋白质折叠[58-61],该领域的多个方面已经彻底革新。蛋白质设计和结构预测相辅相成,设计过程可以看作是预测过程的逆向工程。蛋白质设计可以利用结构特征来指导设计任务,而结构预测技术的进步为设计工作提供了实用的工具,从而推动了蛋白质研究的发展。蛋白质序列中包含了其折叠结构的关键信息,深度学习技术的应用加深了研究人员对序列与结构关系的理解,为基于结构的蛋白质设计奠定了坚实的基础。
在固定骨架的情况下,寻找最大化氨基酸联合概率的序列是设计的关键[62]。传统设计方法倾向于产出与输入骨架紧密匹配的序列,忽略了结构的灵活性与动态性,导致输出限制和序列多样性不足。然而,深度学习能够从数据中学习到人类所不知道的潜在规律,有望解决上述局限性。
2018年,一种名为SPIN2的新模型被提出[63]。最初,SPIN2模型的平均恢复率为34.0%,它仅采用了一维属性。随后,采用了基于图像学习框架新方法[64],在独立的测试集上,序列的恢复率提高到39.8%。进入2020年,进一步开创了DenseCPD深度神经网络[65],这是一种考虑蛋白骨架原子三维密度分布的模型,其在两个测试集上氨基酸回收率分别达到55.53%和50.71%,比以前最先进的技术提高了约10.00%。
TrDesign[66]以TrRosetta[58]模型为基础进行反向序列设计,利用比对蛋白序列的几何特性,以推测残基间的距离分布图。接着通过蒙特卡罗模拟退火技术,对随机序列进行迭代优化;在这个过程中,可以在任意位置突变氨基酸,当新序列的距离分布满足Metropolis标准时接受这次突变,完成所有替换后将创造自然界从来没出现过的序列。然而,模型的反复运行可能会降低计算效率,陷入次优解的方案。刘海燕课题组开发了ABACUS-R[39-40,67],使用Transformer神经网络,多任务学习策略,消除重构和优化侧链结构的需要,简化序列设计的过程,使得平均恢复率提高到53%。生物实验验证表明,ABACUS-R的设计精度和成功率均超过现有最先进的能量函数方法。
2022年,David Baker团队提出了一种基于深度学习的蛋白质序列设计方法[29],称为ProteinMPNN (图4),通过不同采样温度和噪声添加,旨在增强序列多样性及可靠性,该方法在模拟和实验中表现卓越。与基于物理的方法如Rosetta不同,ProteinMPNN无需为每一个特定的设计挑战进行专家级定制,可以广泛应用于多种序列设计问题中。这种广泛适用性源于序列设计构建方式的本质区别,并依赖于通过从PDB检索所有蛋白质进行直接训练的深度学习方法,而不是识别最低能量氨基酸序列问题。由于具有较高的实验设计成功率和计算效率,ProteinMPNN有望成为蛋白质序列设计的标准方法,并可能迅速获得研究人员的广泛认可。此外,它还展现出更高的结晶倾向性,极大地促进了设计蛋白质结构测定。预计ProteinMPNN生成的序列将大幅改善天然蛋白质骨架的稳定性和表达能力,对于重组表达的天然蛋白质尤为有益。由于设计方法的最终检验是实验验证,就像翻译的准确性需要人来评估一样,ProteinMPNN的实用性同样需要通过实验来验证。
2023年,一种基于深度多层感知机的前馈神经网络Anand模型[68]被提出,该模型通过16个氨基酸骨架原子定义残基的结构环境。实验证明,该方法能够精准地预测折叠序列,并产生与参考序列差异显著的新氨基酸序列,这为蛋白设计和材料科学提供了更大的自由度。
随着蛋白质结构数据日益丰富和深度学习技术的不断进步,基于结构的序列设计方法层出不穷,这些方法的恢复率和预测精度都在持续提高。不过大部分蛋白设计工作结构类型不确定,因此可变骨架设计在设计蛋白质时需要优化同时序列和结构。深度学习技术通过识别结构的模式,可以极大地提高这些模式的辨识度并输出增强的特征。
图4 ProteinMPNN模型的整体结构
2021年,一个新颖的可变骨架蛋白质设计技术[69]被提出,该技术是训练一个可以迭代优化序列的网络,被称为“幻想(hallucination)设计”。该方法使用TrRosetta来预测序列的空间约束,并利用Kullback-Leibler散度进行迭代优化,使得预测的结构更加接近真实蛋白的空间分布。在TrDesign-motif[70]的应用案例中,它结合TrRosetta和幻想设计,通过优化目标分布与背景噪声分布的结构和序列,专门设计活性位点和功能motif。RFDesign则通过“constrained hallucination”来优化蛋白质设计[71]。“Inpainting”方法使用RoseTTAFold[18]在补全方面作出了创新,它能够同时生成序列和相应的结构。这些方法已应用于创建结构上有效且具有潜在生物活性的金属蛋白和酶等,且通过实验验证。然而,RFDesign在使用RoseTTAFold时面临挑战,受限于单次的运行预测结构。为了提升蛋白质从头设计的质量和多样性,新工具AutoFoldFinder[72]采用CM-Align进行优化设计,这一方法优化了序列并生成结构,克服了传统KL散度方法的局限,大幅提高了新蛋白质结构的比例,同时显著提升了从头设计的质量,产生了与现有结构差异显著的蛋白质。
在近期的研究中,扩散模型在蛋白质设计领域的应用展现出显著的潜力。2022年,有研究者开发了使用等变去噪扩散概率模型(equivariant denoising diffusion probabilistic models)来生成蛋白质结构和序列的方法[73]。该方法能够从实验数据中学习并生成具有特定三维结构和化学性质的蛋白质,以实现目标功能。紧接着,2023年,研究人员对RoseTTAFold结构预测网络进行微调[20],将其应用于蛋白质结构去除噪声任务,并开发出一种名为RFdiffusion的蛋白质骨架的生成模型;该模型在多个领域表现出色,包括无条件蛋白质单体设计、蛋白质结合剂设计、对称寡聚体设计、酶活性位点和对称基序支架设计等,特别适用于解决蛋白质设计中的复杂问题,如从头设计结合蛋白、设计具有特定对称性的蛋白质结构以及创建新的蛋白质药物和疫苗;该团队使用了来自蛋白质数据库(protein database, PDB)的结构为基础,并对其施加多至200步的噪声,以此作为训练输入;RFdiffusion模型利用最小化的预测与未对齐的真实蛋白质结构之间的均方误差损失进行训练,实现噪声过程逐步逆转;在序列设计方面,采用ProteinMPNN对结构所对应的序列进行编码,通常对每个生成8个序列变体;尽管RFdiffusion在结构和序列设计方面都具有潜力,可是由于其与ProteinMPNN单独结合已显示出良好的表现,所以并未广泛探索这种潜力。研究者通过实验验证了数百种设计的对称组装体、金属结合蛋白及蛋白质结合剂的结构和功能,证实了RoseTTAFold扩散方法的强大功能和广泛适用性[74]。其中,RFdiffusion作为一种前沿的蛋白质设计技术,在实验层面已成功构建了多样的蛋白质结构,包括具有创新性拓扑特征的蛋白质、对称性寡聚体、酶活性位点的支架以及金属结合位点的支架。该技术能够设计出具有独特α/β桶状结构的蛋白质,并能精确地设计出与特定靶标如SARS-CoV-2病毒的刺突蛋白相结合的作用蛋白。此外,RFdiffusion还能够设计出具有特定对称性,例如C3对称性的三聚体结构,这些设计在电子显微镜下展现出与设计模型高度一致的结构,从而证明了其在创新蛋白质结构生成方面的准确性和潜在应用价值。尽管RFdiffusion在蛋白质设计领域展现出显著优势,能够创造出具有新颖结构和功能的蛋白质,并且其设计的蛋白质在实验中被证实具有预期的结构和功能,但也存在一些局限性。首先,设计复杂性受到现有计算资源和算法效率的限制,对于大型蛋白质结构的预测可能超出了单序列预测的能力。此外,设计过程可能需要结合其他工具以确保序列的稳定性;对称性设计的限制需要进一步地优化,以确保蛋白质在生物体内的结构稳定性和功能。同时,在实际应用中还需要克服包括外部条件依赖性、生物活性和免疫原性的考量、知识产权和伦理问题、成本和可及性以及软件和算法的局限性等障碍。尽管面临这些挑战,RFdiffusion在蛋白质设计领域的应用前景仍然非常广阔,尤其是在药物开发和生物技术领域,它为解决复杂的设计挑战提供了新的可能。
2024年,David Baker团队在蛋白质设计领域作出了显著贡献,他们提出了一种名为全原子模型(RoseTTAFold All-Atom, RFAA)的先进计算方法[74]。RFAA通过在去噪任务上的精细调整,进一步发展为RFdiffusionAA模型,该模型在多种应用场景中展现出广泛的适用性,包括药物设计、蛋白质工程和生物标志物的识别。RFAA模型特别擅长设计能够与特定生物标志物特异性结合并调节其功能的蛋白质,以及设计能够靶向并破坏病原体(如病毒或细菌)的蛋白质。例如,该模型已被用于围绕小分子构建蛋白质结构,并通过晶体学和结合测量实验验证了其设计的蛋白质与心脏疾病治疗剂地高辛、酶辅因子血红素和光捕获分子胆红素的结合,显示出高特异性和稳定性。RFAA的主要优势在于能够处理广泛的生物分子复合体[74],并在预测蛋白质-小分子复合体结构方面展现出高精度。然而,RFAA的性能在很大程度上依赖于训练数据集的质量和多样性。如果训练数据集中缺少特定类型的生物分子或蛋白质-小分子相互作用,RFAA在预测这些特定情况时可能会受到限制。此外,作为一个复杂的深度学习模型,RFAA的训练和运行需要大量的计算资源,这可能限制了其在资源有限的环境中的应用。在处理与训练数据集差异较大的新蛋白质或小分子时,RFAA的预测准确性可能也存在局限。虽然RFAA旨在模拟广泛的生物分子组装体,但在特定的应用场景中,可能需要更特化的模型来提高预测精度。将RFAA的预测转化为实际的蛋白质设计和实验验证也面临一系列挑战,包括实验条件的优化、蛋白质表达和纯化等。尽管存在这些限制,RFAA在药物发现和生物分子设计领域仍具有广泛的应用前景,特别是在需要精确模拟和设计蛋白质-小分子相互作用的场景中。
在同期的研究中,该实验室利用经过微调的RFdiffusion网络,成功设计了针对特定抗原表位的抗体。这些抗体经过冷冻电镜解析,证实与设计结果高度一致,能够特异性地结合指定的表位[75]。该研究的应用主要集中在快速开发抗体药物和个性化医疗领域。例如,在面对新出现的病毒如SARS-CoV-2时,传统的抗体发现方法可能耗时数月甚至数年,而该研究展示的从头设计方法能在几周内设计出与病毒表面特定蛋白(例如受体结合域)结合的抗体。这些抗体设计具有高亲和力和特异性,能有效中和病毒,阻止其感染细胞。研究中设计的抗体与流感血凝素的结合,冷冻电镜结构与设计模型高度一致,显示出原子级的精确度;抗体与流感的结合亲和力达到了78 nmol,尽管这一亲和力相对较低,但在未经优化的情况下,该结果已经证明了设计方法的有效性;此外,抗体与目标表位的结合显示出高度特异性,如针对TcdB细菌毒素的抗体设计在实验中显示出特异性结合,而对结构相似的TcsL细菌毒素则没有结合活性[75]。总体而言,该研究提供了一种快速且精确的新型抗体设计方法。其创新之处在于利用RFdiffusion网络和RoseTTAFold2结构预测网络,实现了从头设计抗体的突破,为抗体药物的快速开发提供了新的可能。然而,目前设计的抗体与目标的亲和力相对较低,设计成功率也有提升空间。该方法适用于需要特定表位结合的抗体设计,特别是在缺乏天然抗体或需要快速开发新型抗体药物的情况下。此外,该方法的通用性可能适用于多种疾病相关抗原,为个性化医疗和精准治疗提供了新的工具,标志着结构引导的大分子抗体设计领域的一个新起点。这不仅为抗体药物的快速和成本效益高的开发开辟了新途径,也为扩散模型在生物工程中的进一步应用奠定了基础。
在国内,刘海燕研究团队提出了一个名为SCUBA的全新方法[76],该方法使用的是一种神经网络形式的能量项统计模型,实现了基于连续采样和优化主链能量的新的蛋白主链骨架设计。SCUBA模型融合了局部构象倾向性、氢键几何构形以及侧链所需骨架空间等关键因素,利用邻接计数和神经网络方法进行训练,以此生成全新设计的蛋白质主链骨架;并且采用随机动力学和模拟退火算法,结合之前提到的ABACUS2技术进行序列优化和骨架设计的迭代过程,实现了可变骨架蛋白的从头设计。研究表明,SCUBA设计的骨架不仅具有比自然蛋白结构更高的热稳定性,而且序列同一性低,其中约42%的设计蛋白质能够实现正确的折叠[76]。同期另一项研究介绍了SCUBA-D[77],这是一种通过去除噪声扩散的方法,该方法可以从噪声较大的原始骨架中生成高质量骨架。SCUBA-D的创新特点是引入了基于序列语言模型的扩散辅助和若干生成对抗性网络(generative adversarial networks, GAN)式判别器,这些工具共同增强了产生骨架的物理合理性。SCUBA-D的效果可以通过在生成的骨架上设计氨基酸序列并利用结构预测进行评估。这些工具还有一定的局限性,例如无法设计大分子蛋白质且成功率比较低、应用场景较小。
综上,在深度学习领域,通过构建能量函数来捕捉骨架结构与序列之间的关键特征已成为一种有效的策略。与传统的分子模拟方法相比,深度学习在模拟范德华力、氢键、亲疏水性以及其他相互作用关系方面展现出了显著的优势。特别是扩散模型的引入不仅显著提高了预测的准确性,而且为设计具有特定功能的蛋白质提供了一种强大的工具,从而推动了功能蛋白设计领域的进步。此外,深度学习技术的应用促进了蛋白质设计方法的演变,从传统的小蛋白设计逐步过渡到大分子抗体的从头设计,并且在此过程中显著提高了设计的效率。深度学习在基于骨架结构-序列的蛋白质设计方法中扮演着至关重要的角色,为探索已知蛋白质结构空间提供了新的可能。
2.2 不依赖于结构的直接序列生成
如前所述,在蛋白质设计领域,核心目标是找到能稳定展示所需特性并履行其功能的序列。由于蛋白质序列数据较为丰富,且信息流中结构数据过多的转换和中继点可能导致信号偏移,故直接在序列与功能空间建立映射的方法具有潜在优势[78]。相较于在特定骨架上搜索适配度,直接序列设计一旦掌握了序列空间重要分布,便可以指导设计过程,无需依赖预先获得的结构信息。因此深度学习在不依赖结构的直接序列设计方法中展现出强大潜力。
2017年,一种新颖的策略被引入,该方法使用变分自编码器(variational autoencoder, VAE)嵌入天然蛋白质序列[79],能够预测蛋白质突变对功能的影响。利用这种无监督学习技术可以捕捉天然蛋白质的变异,并识别出特定位点间的相互作用模式。与传统的不考虑序列间相互作用的基线方法相比,此策略展现了更高的性能,有时超越了利用Inverse Potts Model[80]的先进技术。作为生成模型,VAE可以指导蛋白质序列空间的探索,从而增强蛋白质设计的合理性和自动化程度。紧接着,一种改进的生成对抗网络模型被提出[81],使用生成的对抗性网络(wasserstein generative adversarial networks, W-GAN)用于生成预测具有特定抗生素抗药性的蛋白质序列。W-GAN模型由于能有效模拟真实数据分布而获得认可,并可生成风格类似于初始训练数据的新数据。然而,上述生成模型在某些方面存在局限性,如生成特定功能的序列时精度不足。2019年,研究人员进一步开发了蛋白质溶解度生成对抗性网络(protein solubility generation adversarial network, ProGAN)的数据增强算法[82],此深度神经网络能从序列预测蛋白质溶解度,并借助ProGAN提高预测的准确性;该研究结果表明,ProGAN生成的数据能提升模型的预测性能相比先前同类研究提高了约10%。
此外,还有研究利用长短期记忆递归神经网络(long short-term memory recurrent neural network, LSTM RNN)单元开发了创新组合和设计多肽序列的新方法[83]。LSTM RNN能捕获顺序数据中的特征,并根据学习到的上下文特征生成新的序列。研究者以氨基酸序列作为输入,针对螺旋抗菌肽的设计模式进行了LSTM的训练,并成功从头生成了82%具有预测抗菌活性的潜在抗菌肽的序列,所生成的序列比随机序列更贴近于训练数据[83]。LSTM RNN作为一种循环神经网络,解决了传统RNN训练中的梯度问题,其循环连接和单元状态帮助保持长期的数据关联。这些技术已成功应用于蛋白质序列分析领域,包括二级结构预测、同源性检测和亚细胞区室定位等。对抗菌肽的系统训练的研究揭示了LSTM RNN在肽和蛋白质设计中的潜能。
2021年,有研究者运用VAE拟合细菌荧光素酶蛋白在序列水平上的分布,并通过解码邻近的潜在载体生成新的变异靶蛋白[84]。它在近70 000个萤光素酶的数据集上经过训练,由条件模型生成的所有23个变体都保留了发光功能[84]。这一结果证实了深度生成模型探索蛋白质可能的序列空间,并成功生成新蛋白的可行性,为传统的合理设计和定向进化方法提供了一个补充策略。2023年,一个新的密集-自动生成的对抗性网络(dense-auto generative adversarial networks, Dense-AutoGAN)模型[85]被提出,该模型融合了注意力机制与GAN,旨在创造出新的蛋白质序列。此模型结合注意力机制和编码器-解码器框架,不仅提升了蛋白质序列的生成质量,还实现了在保留原始特征的同时进行微调。此外,Dense-AutoGAN采用密集连接卷积神经网络的结构,在GAN的生成器中实现了多层的特征传递,既拓展了训练空间,又增强了序列生成的效率。在蛋白质功能图谱上,该模型成功生成了更为复杂性的蛋白质序列。通过与其他模型进行比较,Dense-AutoGAN生成的序列在化学和物理性质上显示出了高准确性和效率,证实了该模型的优越性能。
在蛋白质设计领域,VAE和GAN已被证实可以有效预测和生成具有改进的突变序列。经过模型训练,研究人员能够成功预测实验突变扫描中的突变,并通过实验方式验证了这些序列的功能。随着测序技术的迅猛发展,蛋白质序列数据按照指数级别增加,大量积累了未标记序列。这些序列为大语言模型应用于蛋白质设计领域(表1)奠定了坚实基础。这使得我们能更加深入理解蛋白质序列与功能的关系。
大型语言模型,如ESM-1b大型Transformer模型[87],已成功解析数亿条蛋白质序列,深入挖掘了生物特性。这些模型不仅掌握了蛋白质的二级结构,还成功地捕获了蛋白质多层面的空间组织原则,涵盖了从物理化学属性到远程同源性等多个维度。同样地,在2023年,一种名为ProGen模型[93]被开发出来,这一基于Transformer的语言模型能通过标记不同属性的氨基酸序列进行训练,控制生成带有特定属性且天然蛋白相似的蛋白质。同时,ProtTrans模型[91]在总量为2亿的蛋白质序列上进行了自回归和自动编码器的训练,并通过它展示了无监督语言模型在蛋白质生物物理学特性学习方面的有效性。UniRep模型[86]通过预训练大量未标记的氨基酸序列,有效提取了蛋白质序列的深层特征,并准确预测了天然和从头设计蛋白质的稳定性。借助大数据集预训练可以增强模型的迁移学习能力,进而为特定任务进行微调提供可能。
表1 蛋白质序列大语言模型
2022年,ProtGPT2模型[95]的引入进一步推动了这一领域的发展,这个拥有7.38亿参数的自回归模型,经过Uniref-50数据集的训练;ProtGPT2生成的蛋白质中88%为球形结构,与自然序列相符;对蛋白质数据库的深度搜索结果显示,ProtGPT2序列与自然蛋白序列存在远程同源性;相似性网络分析也表明,ProtGPT2有效地探索了蛋白质功能空间中的新区域。AlphaFold2对ProtGPT2序列进行结构预测生成了具有实验价值和大环特征的良好折叠非理想化结构,展示了新的拓扑种类。此外,这些生成的序列在稳定性和动态特性方面与天然蛋白质序列类似,并在进化序列空间中展现出独特性。在2023年,ProGen2模型[94]被推出,该模型是一种规模更大的自回归Transformer,拥有6.4亿参数,并在超过10亿种蛋白质序列的多样化数据集上进行训练。经过全面评估,ProGen2在蛋白质序列生成、经微调的特殊结构序列以及特定于抗体序列生成方面均表现卓越,突显其在合理序列生成上的优势。2024年,国内之江实验室的科研团队开发了指导蛋白质语言模型(instruct protein language models, InstructPLM)蛋白质设计框架[96],其利用了大型语言模型的跨模态对齐和指令微调技术来指导蛋白质语言模型生成符合特定结构要求的蛋白质序列。这种方法特别适用于需要设计具有特定功能和结构的蛋白质的场景,例如药物开发、生物技术应用和材料科学。经实验验证,成功从头设计具有所需催化特性的酶、结合特性的蛋白质序列、特定结构和功能的蛋白质基材料等。InstructPLM提供了一种强大的工具,用于设计具有特定功能和结构的蛋白质,这在药物开发、生物技术和材料科学等领域具有重要的应用价值。
深度学习在特征提取、模式识别和目标生成方面具有先进能力。在蛋白质设计方面,直接从序列出发的方法因其不依赖预先获得的结构及大量序列信息而显示出优势,尤其是基于深度学习的策略,如大语言模型在不依赖结构的序列设计中表现出强大潜力。这些策略与基于结构的蛋白设计不同,它通过学习序列与其结构功能的关系,能够直接探索序列空间,从而带来蛋白质设计范式的创新[97]。
>>>>
3 总结与展望
近几年,深度学习等人工智能技术已使蛋白设计取得质的飞跃。AI模型凭借其强大的数据处理能力和模式识别能力,在蛋白质结构与序列数据分析上作出了突破性的贡献。通过复杂的网络架构和算法,AI能够识别出关键生物学特征,进而构建预测模型以辅助不同的设计场景。例如,某些深度学习模型设计的蛋白质,在实验中验证具备预定的结构和功能。这不仅证明了AI在蛋白设计中的实用性,也显著提升了设计过程的效率。
尽管AI在这一领域取得了一定进展,但仍存在问题和挑战。首先,蛋白质结构数据库数据规模不足,限制了模型的准确性和泛化能力。这些不足可能通过数据增强、迁移学习等方法得到缓解。其次,对蛋白设计模型性能的现行评估体系,如天然序列恢复率和预测结构之间的差异[52],未能充分反映蛋白设计的物理化学复杂性,缺乏严格和标准化的基准。另外,蛋白质功能的实现往往涉及到动态过程,现有模型通常只关注单一功能状态,而较少综合考虑表达性、溶解性、稳定性和免疫原性等多样属性。最重要的是,实验验证仍是确认AI在蛋白质设计中的关键一环。
随着计算能力增强、算法优化以及新一代高通量实验技术的涌现,研究人员将获得更全面和更深入的生物数据。这将使AI模型的训练更充分,加深对生命过程的洞察,推动设计更复杂精准的蛋白质,使得深度学习模型能够更有效处理复杂的生物数据。未来,人工智能将整合表观遗传学、转录组学和蛋白质组学等生物信息,更准确地模拟生物分子交互,设计出具有特定功能的蛋白质。此外,厘清深度学习模型的“黑箱”机制,增强模型的精确性和计算过程的可解释性[97],将提高药物设计在酶工程、合成生物学以及蛋白工程等领域中的成功率。
最后,随着蛋白质设计的要求越来越个性化和精准化,定制功能蛋白质将成为新常态。人工智能技术将在预测和设计特异性功能、提高设计效率、优化生物合成路径等方面发挥核心作用。这些技术的进步将推动蛋白质设计在个性化医疗、疾病治疗和生物制造等宽广领域的革新,并开启蛋白质从头设计的新时代。
「推荐阅读 」
☆机器学习在蛋白质功能预测领域的研究进展
☆基于深度学习的蛋白质建模与设计
☆抗菌肽结构改造与人工智能研发策略
更多内容,详见最下方【阅读原文】
关于本刊
《生物工程学报》(1985年创刊,月刊)是由中国科学院微生物研究所和中国微生物学会共同主办,国内外公开发行的学术期刊。本刊重点报道生物工程领域的最新研究成果与进展,主要栏目有:综述、工业生物技术、合成生物技术、环境生物技术、农业生物技术、食品生物技术、医药生物技术等。《生物工程学报》是北大中文核心期刊、中信所中国科技核心期刊、中国科学引文数据库CSCD核心期刊、中国科技期刊卓越行动计划入选期刊。本刊已被美国医学索引MEDLINE/PubMed、荷兰Scopus、美国化学文摘CA、美国生物学文摘BA、俄罗斯文摘AJ、日本科学技术社数据库JST、英国国际农业与生物科学研究文摘CABI、美国剑桥科学文摘(自然科学卷)CSA(NS)、波兰哥白尼索引IC、荷兰医学文摘EMBASE、中国科学引文数据库(CSCD)、中国知网(CNKI)、万方数据库等国内外多个检索数据库收录。《生物工程学报》曾获得百种中国杰出学术期刊(2012年)、中国精品科技期刊(2014−2017年)、中国科学院科学出版基金科技期刊排行榜三等奖(2015−2018年)等荣誉,并获得中国科协精品科技期刊学术质量提升项目(2015−2017年)、中国科协中文科技期刊精品建设计划“学术创新引领项目”(2018年)、连续两期中国科技期刊卓越行动计划项目(2019−2023年,2024−2028年)资助。
期刊订阅:
1.直接联系联合编辑部订购(订购全年期刊可享9折优惠,当期作者购刊可享5折优惠),发行部 E-mail:bjb@im.ac.cn ;Tel:010-64807336;
2.各地邮局订阅:邮发代号82-13;
3.网上购买:搜淘宝店、微店店铺名称:中科期刊(订阅及销售过刊);科学出版社期刊发行部:联系电话010-64017032 64017539;
或扫描下方二维码:
——中文期刊联合编辑部出品
疫苗
2024-12-13
摘要:免疫疗法开启了癌症治疗的新时代,但癌症仍然是全球死亡的主要原因。在各种治疗策略中,癌症疫苗通过激活免疫系统特异性地针对癌细胞显示出了希望。尽管当前的癌症疫苗主要是预防性的,但在针对肿瘤相关抗原(TAAs)和新抗原的靶向方面的进步为治疗性疫苗铺平了道路。将人工智能(AI)整合到癌症疫苗开发中正在彻底改变这一领域,通过增强设计和交付的各个方面。本综述探讨了AI如何促进精确表位设计,优化mRNA和DNA疫苗指令,并通过预测患者反应实现个性化疫苗策略。通过利用AI技术,研究人员可以导航复杂的生物数据集并发现新的治疗靶点,从而提高癌症疫苗的精确性和有效性。尽管AI驱动的癌症疫苗显示出前景,但仍存在重大挑战,如肿瘤异质性和遗传变异性,这可能限制新抗原预测的有效性。此外,必须解决围绕数据隐私和算法偏见的伦理和监管问题,以确保负责任地部署AI。癌症疫苗开发的未来在于将AI无缝整合,创造个性化免疫疗法,提供针对性和有效的癌症治疗。本综述强调了跨学科合作和创新在克服这些挑战和推进癌症疫苗开发中的重要性。
1.引言
癌症是全球死亡的主要原因,在2020年大约造成了1000万人死亡。作为全球主要的健康挑战之一,它仍然是研究和创新的关键领域。在各种治疗方法中,免疫疗法因其能够利用人体自身的免疫系统对抗癌症而受到显著关注。一个有前景的免疫治疗策略涉及开发癌症疫苗,这些疫苗旨在刺激免疫系统识别和攻击肿瘤细胞。这些疫苗通过靶向肿瘤抗原(TAs)工作,这些是癌细胞上表达的独特蛋白质或分子。通过人为诱导对这些抗原的免疫反应,癌症疫苗旨在对肿瘤产生特异性和持久的免疫力。与传统疗法不同,癌症疫苗提供了更安全、更有针对性和更好耐受性的方法。然而,它们的临床转化面临许多挑战,主要是由于肿瘤抗原的异质性景观和个体免疫反应的固有变异性。尽管已有几种癌症疫苗,如Cervarix、Gardasil、Gardasil-9和乙型肝炎疫苗(HEPLISAV-B),主要用于预防目的,但正在研究的先进疫苗旨在特别针对癌细胞上的标记,如TAAs或新抗原,以激活免疫反应并有效攻击这些肿瘤。AI作为这一领域的变革性工具,显著加速了创新癌症疫苗的开发。大量公共数据的可用性进一步推动了这些进步,为研究人员提供了前所未有的机会来识别和验证新靶点。本综述探讨了AI与癌症疫苗开发之间的动态互动,提供了AI如何通过增强疫苗发现和优化的速度和精确度来彻底改变这一领域的见解。疫苗技术的最新进展,结合对癌症免疫学的更深入理解,为创新的治疗途径铺平了道路。AI在癌症领域的干预是深远和多方面的,对治疗和研究具有重要意义。通过数据驱动的模式识别,AI在检测突变和解析复杂的基因组签名方面发挥了重要作用。现代免疫学和数据科学的整合为疫苗生产引入了创新的分析方法。例如,深度学习模型能够探索基础、转化和临床研究中的广泛可能性,加速开发高效的癌症疫苗。此外,AI可以对癌症疫苗的反应者或非反应者进行分类,实现更个性化的治疗策略,改善患者结果,并为那些对传统疫苗无反应的人提供替代疗法。像IBM的Watson Oncology这样的先锋平台展示了AI在个性化癌症治疗中的潜力,利用庞大的数据存储库提供量身定制的治疗建议。同样,AI在免疫学领域也取得了显著进展,特别是在表位预测这一关键步骤中。这一领域的一个值得注意的工具是DiscoTope-2.0,它通过分析局部几何结构,包括侧链方向和溶剂可及性,计算每个残基的表位倾向。这个工具在预测B细胞表位方面发挥了重要作用,这对于设计有效的疫苗至关重要。在此基础上,DiscoTope-3.0引入了一种更复杂的方法,通过整合正非标记学习技术和创新的逆折叠结构表示。与其前身不同,DiscoTope-3.0是多功能的,适用于预测和实验解决的结构。最重要的是,DiscoTope-3.0克服了以前模型的限制,即使对于放松和预期的结构也能保持高预测准确性,从而消除了对实验结构数据的依赖。这一进步不仅加速了表位映射过程,而且扩大了可以分析的抗原范围,使其成为疫苗研究和开发的强大工具。
有效的癌症疫苗设计之路充满了挑战,从确定最佳靶点到克服先前的免疫耐受性。当考虑到突变抗原、患者特定变异和肿瘤抗原的动态演变时,复杂性加深。随着AI继续其上升趋势,本综述旨在展示AI与癌症疫苗设计过程之间的协同作用。它将复杂的生物学概念变得更容易理解,无论是对生物学家还是计算机专家。最终,本综述开启了癌症疫苗开发的新时代,AI和人类智慧共同应对我们时代最紧迫的健康挑战之一。虽然AI和ML(机器学习)技术可以加快疫苗开发的某些方面,但它们不能取代动物模型和人类试验在确保安全性和有效性方面提供的基本验证。然而,本综述并未涵盖癌症疫苗研究的所有方面。它特别排除了监管挑战、大规模临床试验设计和T细胞激活和免疫记忆形成的详细免疫学机制等主题,这些超出了AI驱动方法的范围。本综述简要讨论了AI在医疗保健中使用的伦理影响,但它们不是本综述的主要焦点。
2.综述方法论
这种周到的安排有助于新手和经验丰富的研究人员探索这一领域。据我们所知,以前没有研究彻底检查过这些技术,特别是AI相关技术与疫苗技术最新进展的协同组合。我们还确定了与基于AI的工具相关的持续挑战,并提出了创新的研究方向来解决这些问题。这样的综合努力可以促进先进AI技术的完善和广泛采用,以开发有效的基于疫苗的癌症疗法。附录部分包括表A1,提供了文章中使用的缩写词汇表。
2.1.搜索策略和文献来源
图1显示了使用特定关键词进行的数据库查询。本综述遵循严格的文献综述方法,呈现了研究和综述论文的关键发现,探索了机器学习和深度学习技术在基于癌症的疫苗疗法中的应用。采用PRISMA-ScR方法,文章从包括谷歌学术、Medline、PubMed Central、Cochrane Library和Scopus等知名在线数据库中精心挑选。文献广泛记录了这些工具在表位设计、突变预测和各种核酸疫苗设计中的AI整合等不同领域的应用。这一日益增长的证据突出了AI技术在医疗保健中的不断扩大的认可,为实践和研究的关键领域提供了宝贵的见解和重大进展。本综述进一步分析了其他学者的贡献,并为未来的研究工作提出了富有洞见的方向。
图 1、使用特定关键词在数据库中进行的查询。
2.2.纳入标准
文章选择过程限制在英文出版物,优先考虑综述主题的新颖性和相关性。特别关注疫苗,我们精心策划了全面使用机器学习和深度学习(DL)方法在癌症疗法中的文章。只考虑同行评审的论文,另外要求提供原始数据或分析。
2.3.排除标准
排除过程从筛选摘要开始,然后进行数据提取和全文的综合分析。根据多个标准排除文章,包括写作质量差、非英语语言或缺乏相关性。重复出版物和与研究主题无关的也被排除在外。我们也没有包括那些被认为是掠夺性期刊的论文。
2.4.结果
从各种文学来源、Scopus、Medline、PubMed Central、Cochrane Library和谷歌学术获得了785篇出版物。在筛选标题和摘要后,有226篇论文被排除。在彻底审查剩余358篇出版物的全文后,我们进一步缩小了选择范围。最终选择了210篇论文进行进一步分析。这个选择过程的结果在图2中说明。
图 2、基于PRISMA-ScR指南选择研究以纳入综述的方法论。
3.传统疫苗设计过程
治疗性癌症疫苗(TCVs)旨在控制肿瘤生长、消除残留疾病并诱导已建立的肿瘤退缩。图3所示的传统疫苗设计过程突出了创造有效癌症疫苗所涉及的关键步骤。这一过程的核心是将抗原有效地传递给树突状细胞(DCs),这导致它们的激活,随后引发强烈的免疫反应,包括CD4+ T辅助细胞和细胞毒性T淋巴细胞(CTLs)。
图 3、传统疫苗设计流程。疫苗开发之旅始于识别和选择目标抗原,然后将这些抗原与合适的佐剂结合。在进入临床试验之前进行临床前测试。在临床试验中评估疫苗的效力和安全性后,寻求大规模生产和分销的监管批准。
许多疫苗特别配制有病原体相关分子模式(PAMPs),即病原体的成分,被树突状细胞表面的模式识别受体(PRRs)识别。这种相互作用是启动免疫反应的关键。图4进一步说明了TCVs的作用机制(MOA),其中PAMPs与PRRs的结合引发细胞内信号级联反应。这个级联反应通过诱导细胞因子释放和提高它们表面共刺激分子的表达来激活DCs。
图 4、疫苗的作用机制(MOA)。癌症疫苗向免疫系统引入肿瘤相关抗原或肿瘤特异性抗原。抗原呈递细胞(APCs),如树突状细胞,处理这些抗原并以人类白细胞抗原(HLA)限制性方式呈递给T细胞。激活的T细胞识别并结合到表达相同抗原的肿瘤细胞,导致细胞毒性T细胞(CD8+ T细胞)和辅助T细胞(CD4+ T细胞)的激活。CD8+ T细胞直接靶向并杀死肿瘤细胞,而CD4+ T细胞协助其他免疫反应。激活的B细胞产生抗体,中和肿瘤细胞或其分泌的因子,促进肿瘤细胞死亡。T细胞和B细胞的免疫监视监测并清除任何可能逃脱初次治疗的剩余肿瘤细胞。免疫系统保留了肿瘤抗原的记忆,允许如果肿瘤复发,可以迅速响应。此外,癌症疫苗通常与其他免疫疗法或标准治疗结合使用,以增强疗效。
一旦激活,树突状细胞就会经历一个成熟过程,增强它们向T细胞呈现抗原的能力。图4详细说明了这种成熟,涉及表面分子的变化,特别是MHC分子和共刺激分子如CD80和CD86的表达增加。然后DCs将抗原处理成肽片段,这些片段在它们表面的MHC分子上呈现。成熟后,DCs从抗原遭遇地(如疫苗接种点)迁移到附近的淋巴结。在这些淋巴结中,DCs与原始T细胞相互作用,呈现抗原-MHC复合物以及共刺激信号,这导致T细胞的激活。如图4所示,CD4+ T细胞分化为辅助T细胞,而CD8+ T细胞成为细胞毒性T细胞。辅助T细胞刺激B细胞产生抗体,而细胞毒性T细胞直接攻击和消除受感染或癌细胞。
疫苗的给药方式对其效果和安全性起着至关重要的作用。虽然肌肉注射是最常用的途径,被认为是诱导全身免疫的黄金标准,但它在提供粘膜保护方面存在局限性。某些疫苗也倾向于使用皮内和鼻内给药方法,因为它们可以直接接触到皮肤或粘膜组织中的树突状细胞(DCs),增强这些关键部位的免疫反应。皮内注射虽然需要专门的技术,但效率很高,并且允许使用较小剂量的疫苗。鼻内给药直接针对粘膜表面,促进局部和全身免疫反应,但通常需要添加佐剂以增强效果。皮下注射给药方便,但在刺激强烈的免疫反应方面可能效率较低。口服疫苗虽然方便,但面临消化系统中的降解和免疫原性降低等挑战。了解每种给药途径的优势和局限性对于优化疫苗设计和输送策略至关重要。选择最合适的方法可以显著影响疫苗对抗各种病原体的能力,并确保个体得到强有力的保护。
为了增强疫苗中的T细胞反应,可以采用几种策略,因为更强烈和持久的免疫反应对于有效的癌症免疫疗法至关重要。一个关键方法涉及使用佐剂,这些物质通过刺激抗原呈递细胞(APCs),如树突状细胞(DCs),促进细胞因子的产生,从而增强抗原的免疫原性。除了佐剂外,针对特定树突状细胞受体也是一个有前景的新策略。这种方法侧重于通过专门设计的抗体将抗原局部输送到表达在DCs表面的内吞受体上。DC基疫苗中最有希望的靶标之一是DEC-205,这是一种DC特异性的内吞受体,能有效内化抗原并将它们导向MHC II类途径。临床前模型已经证明,用抗原特异性抗体靶向DEC-205可以引发强烈且持久的体液和细胞免疫反应。此外,其他DC特异性受体,如Clec9A和Clec12A,也显示出作为增强DC基疫苗的靶标的潜力。通过选择性靶向这些受体,疫苗可以更有效地激活免疫系统,导致更强的T细胞反应。
此外,通过靶向特定的DC亚群来定制免疫反应可以进一步细化结果,引导免疫系统朝着更理想和有效的反应方向发展。一个重要的考虑因素是肿瘤微环境(TME),在其中持续的免疫细胞浸润和长期维持免疫反应对于成功控制肿瘤至关重要。一种策略涉及使用载有来自TME中死亡肿瘤细胞的肿瘤抗原的自体DCs。然而,这些方法面临几个挑战,包括肿瘤异质性,肿瘤的不同区域可能表达不同的抗原,可能导致不完整的免疫反应。此外,复杂抗原的呈现不足可能导致T细胞激活不足,限制了整体治疗效果。还有风险是免疫系统可能不会对肿瘤细胞发起足够强烈的攻击,因为它可能将肿瘤抗原视为自身抗原,从而降低免疫反应。因此,癌症疫苗的设计和开发是一个复杂和多步骤的过程,需要仔细考虑。这包括识别特定的癌症抗原,评估它们的免疫原性,用适当的佐剂配制疫苗,并进行全面的临床前和临床测试。该过程经历了多个阶段的安全性和有效性评估,最终导致监管批准和大规模生产。
4.AI在疫苗开发的各个步骤中的应用
AI可以在特征提取和模型训练中协助预测患者特定的癌症抗原。通过复杂的算法,AI可以优化和改进这些抗原,指导疫苗配方,并支持临床试验设计,有可能实现更加个性化的疫苗接种策略。实时监测和持续学习可以确保适应性治疗策略,而遵守监管标准和实验验证可能有助于评估安全性和有效性。随着AI驱动的癌症疫苗开发不断进步,确保这些技术的安全性、有效性和伦理使用需要谨慎地应对复杂的监管环境。
关键挑战可能包括确保AI算法不引入意外的风险或偏见,在整个开发过程中保持透明度,并解决训练数据中潜在的偏见问题。强大的数据安全和隐私措施,以及设计良好的临床试验,对于获得监管批准和维护公众信任可能至关重要。此外,与全球监管机构和利益相关者的合作可能有助于建立协调一致的标准,确保像知情同意这样的伦理考虑得到妥善处理。这种合作最终可能为AI驱动疫苗安全、负责任地融入癌症治疗铺平道路。
癌症疫苗设计的AI模型开发严重依赖于获取广泛和高质量的数据集。表1汇总了可能有助于创建新型癌症疫苗的各种数据集和数据库。许多列出的数据库是表位数据库,如IEDB和SYFPEITHI,以及新抗原肽数据库如dbPepNeo2,和MHC结合物数据库如MHCBN。这些资源可以提供有关癌症疫苗目标抗原和能够与之结合的MHC分子的关键信息,支持研究人员识别有前景的疫苗候选物。
4.1.利用人工智能进行表位设计和主要组织相容性复合体结合预测
表位是抗原上的一个特定分子结构或区域,免疫系统识别它并需要它来启动免疫反应。精心设计表位是靶向治疗中的关键步骤,对于最小化对健康细胞的潜在伤害至关重要。人工智能算法在这一设计过程中扮演着重要角色。人工智能驱动的方法通过考虑相邻氨基酸的相对亲和力,显著提高了预测TCVs设计中表位的准确性。在过去几十年中,研究人员开发了许多用于表位设计和MHC肽结合预测的人工智能算法,如表2所示。由于指标多样,它们在补充论文中进行了详细阐述。它列举了人工智能与癌症疫苗开发交叉点的各种研究。这些研究采用各种人工智能技术,如神经网络(例如,BepiPred和MHCflurry-2)、支持向量机(SVM)(例如,DeepImuno和Epitopia)以及自然语言处理(例如,MHCSeqNet),来预测和评估癌症疫苗的关键方面,包括表位结合、免疫原性和抗原呈递。虽然每个工具都有其独特的优势和局限性,但总体而言,它们强调了人工智能在增强我们对癌症免疫学的理解以及寻找有效的新抗原用于TCVs开发中的变革潜力。
识别新抗原的一个挑战是预测能够与MHC I类分子结合并在黑色素瘤细胞表面呈现的肽。为了解决这个问题,已经采用了多种计算技术,使用机器学习(ML)算法和质谱(MS)数据。基于ML的方法依赖于已知MHC I类结合肽的大型训练数据集,这些数据集通常有限且不完整。因此,这些方法可能会遗漏数据库中没有很好代表的一些新抗原。为了克服这个限制,Abelin等人使用MS对表达单一HLA-I等位基因的细胞的MHC配体组进行分析,并识别了这些等位基因呈现的24,000个肽。他们还检查了蛋白质切割和基因表达水平对抗原呈递的影响。基于MS的方法也可以用来创建更准确和全面的训练数据集,供基于ML的方法使用。例如,一种最近的方法使用MS从人类肿瘤活检中收集了185,000个高质量的MHC I类结合肽,并用它们训练了一个ML模型,实现了超过75%的预测准确率。另一种方法是使用不同的ML模型联盟来提高预测性能。Racle等人创建了MoDec,这是一种类似于卷积神经网络(CNN)的基序解卷积算法,使用基于质谱的肽数据集。这个算法旨在通过结合核心偏移偏好和肽切割基序来识别MHC II结合基序。他们有一个包含23个不同样本和总共77,189个独特肽的数据集。这些方法展示了ML和MS在增强新抗原识别中的协同作用,可能导致更有效和个性化的癌症免疫疗法。
疫苗开发严重依赖于T细胞表位的计算预测,重点关注抗原处理和呈递机制,如与抗原处理相关的转运蛋白(TAP)、蛋白酶体切割和MHC结合。开发准确、快速且能够提供全面洞察这种结合的计算技术可以显著加速免疫疗法和疫苗开发的进展。针对黑色素瘤细胞的一个关键步骤是将这些MHC分子与表位结合并呈递给细胞毒性T细胞。在这种情况下,Chu等人介绍了TransMut框架,包括用于pHLA结合预测的TransPHLA和用于修饰肽优化的自适应优化突变肽(AOMP)。TransPHLA由源自Transformers的自注意力模型驱动,在预测pHLA结合、新抗原和人乳头瘤病毒疫苗(HPV)识别方面表现出色,超越了其他13种方法。
支持MHC肽结合预测和表位设计最常用的技术之一是支持向量机(SVMs)。它们被用来通过利用数据描述来预测T细胞表位,确定肽结合偏好。例如,Zhao等人开发了一个SVM模型用于肽-MHC分子相互作用。开发新型免疫诊断试剂和疫苗的基础是识别抗体优先识别的蛋白质表面区域(抗原表位)。计算技术为抗原表位识别提供了必不可少的工具来支持这一努力。三肽相似性和倾向性得分被结合起来使用SVMTriP。使用五折交叉验证,SVMTriP在应用于从IEDB检索到的非冗余B细胞线性表位时,获得了80.1%的灵敏度和55.2%的精确度。AUC(曲线下面积)结果为0.702。通过结合三肽序列的相似性和倾向性,可以提高线性B细胞表位的准确性。Nagpal等人开发了一种基于SVM的技术来预测肽改变APCs进行抗原呈递的能力。他们开发的调节剂在训练数据集上展示了95.71%的惊人准确率,并且已经通过名为VaxinPAD的基于网络的服务器公开访问。这些技术的有效性强调了持续创新和计算技术精度的重要性,以促进该领域的发展。准确预测结合MHC I类分子并随后在黑色素瘤细胞上表达的肽使用AI方法面临几个重大挑战。首先,实验数据的数量有限和变异性,特别是对于罕见的MHC等位基因,阻碍了AI模型的训练和有效性。其次,人类群体中MHC等位基因的巨大多样性,每个都有独特的结合偏好,需要能够概括这种复杂性的泛特异性模型。进一步加剧问题的是由各种细胞因素影响的肽处理和呈递的复杂和动态性质。最后,从众多可用选项中选择最合适的AI模型需要仔细考虑数据类型、特征、参数和性能指标,以确保最佳的预测准确性。解决这些挑战对于充分利用AI在开发针对黑色素瘤的有效免疫治疗策略中的全部潜力至关重要。
4.2.AI驱动的癌症诊断和个性化治疗策略的进步
生物标志物是用于评估各种生物过程的可测量信号或指标,包括癌细胞的存在或特定治疗的有效性。AI驱动的癌症疫苗的一个关键方面涉及突变和生物标志物预测,使识别特定遗传改变和生物标志物以准确诊断癌症和个性化治疗成为可能。癌症生物标志物是由患有癌症的个体体内或肿瘤内产生的生物分子。这些生物标志物包括各种形式,包括DNA、RNA、蛋白质或代谢模式,并且是肿瘤特有的。通过仔细检查患者特定肿瘤固有的独特遗传或分子改变,医疗从业者可以精确确定最合适的治疗策略。显著地,在以乳腺癌为中心的研究中,HER2生物标志物的识别在识别可以从像曲妥珠单抗这样的靶向治疗中获益的个体中发挥了关键作用。通过HER2生物标志物测试,临床医生可以评估患者癌症对这种特定治疗积极响应的可能性,从而避免无效治疗和可能的副作用。在识别这些生物标志物之后,下一个阶段集中在确定是否有可利用的遗传改变加速肿瘤发展。
这些生物标志物根据其性质和功能被分类为不同的组。遗传生物标志物涉及与癌症相关的特定遗传突变或改变的分析。蛋白质生物标志物涉及与癌症相关的特定蛋白质或蛋白质表达的测量。这些生物标志物可以用来预测治疗反应或监测疾病进展。例如,前列腺特异性抗原(PSA)通常用于监测前列腺癌的治疗反应和检测复发。成像生物标志物利用各种成像技术,如MRI或PET扫描,评估肿瘤特征或治疗反应。这些生物标志物提供了有关肿瘤大小、位置和代谢活动的宝贵信息,有助于治疗计划和评估。液体生物标志物涉及体液中存在的各种物质的分析。这些生物标志物可以提供有关肿瘤特征或治疗反应的非侵入性和实时信息。例如,血液中循环的肿瘤核酸(DNA)可以帮助监测治疗反应和检测最小残留疾病。在这种情况下,Wood等人开发了一个完全自动化的、AI驱动的体细胞突变发现工具,称为Cerebro。该模型采用RF算法评估众多决策树,为潜在突变生成置信度分数。Cerebro在识别验证突变方面表现出色,具有高灵敏度(97%)和阳性预测值(98%)。
异柠檬酸脱氢酶(IDH)基因中的突变是诊断和管理胶质瘤(一种脑肿瘤)的重要生物标志物。使用来自胶质瘤患者的肿瘤切片与苏木精和伊红(H&E)染色,Liu等人建议使用基于GAN的数据增强技术来提高这些突变的预测准确性。通过数据增强,他们的研究极大地提高了预测准确性。在皮肤黑色素瘤的背景下,Bai等人开发了一个有前景的基于mRNA的生物标志物签名。该签名通过Cox比例风险回归和随机生存森林算法的结合创建,优于临床预后标记,并且具有成为皮肤黑色素瘤预测生物标志物的重大潜力。仅基于突变对癌症进行分类是具有挑战性的,因为肿瘤内异质性、低肿瘤纯度和癌症类型之间的常见突变。通过使用现代分析和诊断工具,如多重免疫和遗传分析以及AI,可以更深入地理解个体组织中免疫和其他细胞之间的空间联系。这揭示了相关的肿瘤内异质性,可能对免疫相关和组合疗法产生重大影响。尽管免疫疗法的武器库不断壮大,但它们成功融入临床实践取决于通过学术研究、经济可行性和明显积极的临床结果建立的全面理解。
从替代来源获得的基因组档案,如无细胞DNA(cfDNA),可以提供有关肿瘤遗传景观的宝贵见解,并有助于癌症的分类。机器学习(ML)在分析复杂的基因组数据和提高我们对癌症类型的分类和理解能力方面至关重要。AI预测模型已显示出在识别与各种医疗状况(包括癌症)的成像症状相关的特定突变或遗传特征方面的有希望的潜力。在这种情况下,Mu等人开发了一个基于PET/CT图像的深度学习(DL)模型,以区分携带EGFR突变和野生型非小细胞肺癌(NSCLC)患者,准确率达到0.81。该模型利用放射线特征进行准确预测。DeepVariant使用深度卷积神经网络(DCNNs)在测序数据中寻找小的插入和缺失。在PrecisionFDA Truth Challenge中,它以卓越的准确性超越了其他变异调用器。这项技术可以帮助识别导致癌症的新抗原或插入和缺失。像Fusion-Bloom这样的先进方法通过应用基于转录组组装的结构变异检测技术,提供了改进的融合变异检测。该算法在检测真正的融合变异方面显示出更高的特异性和敏感性。这些基于AI的突变和生物标志物预测工具在推进癌症诊断和个性化治疗策略方面取得了重大进展。这些发展对于提高癌症患者的生活质量具有很大的希望。
4.3.AI在免疫原性预测中的应用
疫苗的免疫原性指的是疫苗能够触发免疫系统启动免疫反应的程度。免疫原性预测有助于简化疫苗开发、降低成本、增强安全评估、优化剂量策略、针对特定人群量身定制疫苗设计以及快速响应。Li等人提出了一种基于序列确定肽免疫潜力的创新方法。他们引入了一种Beta-二项分布方法,并评估了其与其他模型的性能。令人惊讶的是,由于其适应不同大小数据集的能力,卷积神经网络(CNN)成为了最重要的预测模型。此外,该团队还引入了一个独立的生成对抗网络(GAN),称为DeepImmuno-GAN。这种创新方法成功复制了免疫肽,将其物理化学属性和免疫预测与真实抗原对齐。
Diao等人创建了一个名为Seq2NeoCNN的CNN模型,用于预测肽的免疫原性。该模型的性能与其他机器学习模型(SVM、随机森林、ExtraTree、逻辑回归和XGBoost)进行了比较评估,这些模型也使用独立的TESLA数据集进行训练。该模型达到了0.801的准确率。还有许多其他的例子,例如,Wang等人创建了INeo-Epp,这是一个基于随机森林的分类器,用于预测包括新抗原在内的T细胞表位的免疫原性。讨论了特定组织内免疫细胞的总体组成、组织和活性对临床反应的影响,以及机器学习在寻找新的T细胞新表位方面的潜在用途。3T biosciences开发的3T-TRACE平台利用主动机器学习与广泛的目标库一起发现新的目标和T细胞受体(TCR)。这种方法识别了固体肿瘤中最常见和免疫的目标,并适用于所有类型的肿瘤。并非所有MHC呈递的肽都能引发免疫反应。myNEO开发的个性化癌症平台myNEO ImmunoEngine,在预测新抗原的免疫原性时也强调了结构属性。
虽然AI驱动的模型在预测免疫原性方面显示出了显著的希望,但仍存在几个挑战,特别是关于肿瘤异质性和个体遗传变异性。肿瘤异质性——在同一肿瘤内存在具有不同突变负担和免疫逃避策略的不同亚克隆——提出了一个重大障碍。AI模型可能准确预测一个亚克隆的新抗原,但未能考虑到其他亚克隆,导致不完整或无效的免疫反应。这种局限性强调了将更多动态、实时数据整合到AI模型中的必要性,以捕捉不断演变的肿瘤档案。此外,个体遗传变异性,特别是在主要组织相容性复合体(MHC)中,显著影响免疫反应。在常见MHC等位基因上训练的AI模型可能无法很好地推广到具有罕见等位基因的个体,导致某些人群的疫苗设计不够优化。这强调了在包含更广泛遗传背景的多样化数据集上训练AI模型的重要性,从而实现更个性化和有效的疫苗开发。
疫苗过敏性指的是疫苗在接种者中引发过敏反应的潜力。对于过敏性预测,Dimitrov等人引入了AlgPred和AllerTOP 1.0服务器,用于评估嵌合蛋白的过敏性。这些服务器提供了多种确定过敏性的方法,包括使用过敏性蛋白代表性肽(ARPs)和混合方法。这涉及到在Gasteiger等人获得的2890个ARPs数据库中搜索查询蛋白序列。混合选项结合了基于蛋白质一级物理化学属性的无对齐方法和KNN分类,实现了94%的高灵敏度。对于抗原性评估,使用了两个服务器,ANTIGENpro和VaxiJen v2.0(91)。ANTIGENpro是一个无对齐服务器,基于序列,使用蛋白质抗原性质的微阵列数据来预测蛋白质抗原性。相反,VaxiJen v2.0仅基于蛋白质的物理化学特性进行分类,实现了89%的分类准确率。AI在免疫原性预测和相关领域的这些不同的应用展示了AI在推进我们对癌症的理解和治疗方面的广阔潜力。这些研究共同说明了使用AI技术来增强免疫原性预测的精确性和有效性,为更有针对性的和有效的治疗干预铺平了道路。
5.利用人工智能在疫苗设计中的应用
人工智能在癌症疫苗开发中的综合应用是多方面的,涵盖了几个关键过程。首先,人工智能在目标识别和验证中扮演着至关重要的角色。它分析大量的基因组和分子数据,以确定潜在的肿瘤抗原,从而显著优化免疫系统目标的选择。这使得免疫疗法能够更精确、更有针对性。其次,人工智能有助于疫苗的设计和优化。通过模拟与免疫系统的相互作用,人工智能可以预测疫苗的效果,并指导研究人员选择最有前途的候选疫苗进行进一步开发。这显著提高了疫苗创造过程的效率和效果。癌症疫苗具体表现为四种不同类型:基于肿瘤或免疫细胞的、基于病毒载体的、基于肽的和基于核酸的。治疗性癌症疫苗(TCVs)旨在激活适应性免疫系统,协调对特定肿瘤抗原的协同攻击。通过这些机制,TCVs旨在训练免疫系统识别和消除癌细胞,为癌症免疫疗法提供针对性和特定的方法。TCVs通常旨在诱导Th1免疫反应,其特征是激活杀手T细胞和释放促炎细胞因子。这种反应在对抗癌细胞方面特别有效。表3提供了多家公司参与癌症疫苗临床试验的概览,一个共同的主题是它们在开发流程中整合了人工智能技术。这些试验涵盖了多种癌症类型和创新项目。
5.1.人工智能在核酸疫苗设计中的应用
5.1.1.DNA疫苗
这些是由细菌表达的质粒,包含编码抗原蛋白的DNA序列。这些疫苗显示出激发强烈免疫反应的潜力,从而有助于对抗癌症。一个理想的例子是正在进行的针对乳腺癌的临床试验(I期),涉及Mammaglobin-A DNA疫苗(NCT00807781)。另一个例子是针对HPV-16/HPV-18 E6和E7癌基因的DNA疫苗在患有高级别宫颈上皮内瘤变个体中展示的临床效果。DNA疫苗提供了几个优势,包括制造简便、固有佐剂和TAA的良好来源。然而,它们需要额外的转录和翻译步骤,树突细胞才能交叉呈现它们以激活免疫。人们担心DNA疫苗可能整合到宿主基因组中,这可能导致意外的结果,包括激活癌基因或干扰正常细胞功能。免疫系统可能会将DNA载体视为外来物,引发对其的免疫反应。这种反应可能会降低疫苗的效果并引发不良反应。正在努力提高DNA疫苗的免疫原性以提高其效果。选择和微调质粒DNA中的最佳抗原是一种增强疫苗诱导的免疫反应和提高治疗效果的策略。Evaxion Biotech的癌症疫苗EVX-01代表了一种增强的、先进的基于DNA的新抗原癌症免疫疗法,用于转移性黑色素瘤,旨在解决晚期实体癌症。他们的人工智能平台PIONEER有助于为靶向癌症治疗创造新抗原。免疫信息学可以帮助识别可能引发快速和保护性免疫反应的肿瘤特异性抗原,用于DNA疫苗。人工智能算法可以增强这些免疫信息学方法。Lurescia等人讨论了在设计针对B细胞淋巴瘤的DNA疫苗中使用免疫信息学的情况。将人工智能与CRISPR/Cas9基因编辑相结合,开辟了精确基因突变修改、分子克隆和肿瘤基因组改变的新领域。这种协同作用通过利用人工智能的分析能力来解释数据和构建知识模型,加快了基因编辑过程。CRISPR-Cas使得遗传疫苗成为可能,它将某些编码抗原的基因传递到宿主细胞中,以刺激强烈和有针对性的免疫反应。这些基于CRISPR的免疫增强发展对于治疗癌症、传染病和其他有效治疗依赖于强大免疫系统的情况非常有希望。随着这一领域的发展,越来越多的关注集中在增强多表位DNA结构用于临床应用,需要通过实验预测和体外临床前调查彻底验证修改和表位组合。为了最大化效果,必须集中精力在增强表位表达、微调免疫系统的招募和识别最合适的表位组合上。通过利用预测算法、外显子/基因组序列、RNA测序分析以及比较患者肿瘤和标准样本的比较测序,可以确定肿瘤新抗原。可以设计疫苗以触发针对特定识别的新抗原的免疫反应,最小化引发肿瘤生长的风险。Absci,一家生物技术公司,开发了一个名为CO-BERT的密码子优化模型,使用先进的深度学习算法来增强这一过程,特别关注预测实现最大蛋白表达的最有效密码子。这种创新方法允许他们设计具有精确调节的基因序列,以在特定宿主生物体内增强表达,绕过实验室中的传统试错方法。这一进步在DNA疫苗开发等场景中具有重要意义,有助于最佳抗原表达和在体内引发加强的免疫反应。随着持续的进步,基于DNA的癌症疫苗的前景看起来充满希望,能够根据个体MHC/TAA表位配置文件提供定制化治疗。
5.1.2.RNA疫苗
这些通常被称为mRNA疫苗,包含一小段合成的mRNA,编码肿瘤相关抗原(TAAs)或新抗原。与DNA疫苗一样,RNA疫苗具有相对简单的生产优势和固有的佐剂特性。此外,随着对肿瘤突变负担的日益重视和针对新表位的个性化疫苗的潜在益处,在开发癌症治疗的疫苗策略方面取得了显著进展。然而,与DNA疫苗不同,RNA疫苗绕过了转录的需要,允许更快地由MHC分子呈现。这一简化的过程提高了它们在触发免疫反应中的效率。最近的一项研究已经证明了RNA疫苗在激发对癌症的强烈免疫反应方面的潜力,进一步支持了它们作为癌症免疫疗法中的有效策略的潜力。人工智能技术在优化mRNA疫苗结构、增强其免疫原性、安全性和整体效果方面发挥着关键作用。它还可以协助设计自组装mRNA疫苗的纳米颗粒,提高它们的稳定性和传递。
研究人员可以使用深度学习(DL)技术来优化mRNA序列,以增强蛋白表达和抗原呈现,产生更专注和强大的免疫反应。通过机器学习支持的计算机建模,科学家可以模拟免疫系统对癌症等疾病的反应,无论是在有疫苗和无疫苗的情况下。这种方法有助于开发有效的mRNA序列用于疫苗接种目的。了解RNA分子的二级结构对于深入了解它们的细胞功能以及设计新药和疫苗至关重要。在这种情况下,Singh及其同事介绍了一种创新的基于深度学习的预测RNA二级结构的方法。其他工具,如SPOT-RNA、DMfold和CDPfold,专门预测mRNA的稳定性、结构和结合。RNA降解是一个关键过程,影响基因的表达和功能。预测mRNA序列的降解特征,如半衰期和每个核苷酸的降解速率,可以提供对基因表达的调节和动态的见解。然而,由于RNA分子与影响其稳定性的各种因素之间复杂的相互作用,这项任务具有挑战性。He等人提出了一个名为RNA变形器的CNN模型,该模型利用自注意力识别RNA序列中局部和全局依赖关系。卷积层可以提取局部特征,如二级结构,而自注意力层可以学习长距离依赖和相互作用。该模型还通过可视化自注意力层的注意力权重来提供可解释性,这可以揭示不同区域和核苷酸对降解预测的重要性。作者展示了他们的模型在两个数据集上实现了高准确性,并优于现有方法:包含COVID-19疫苗候选mRNA序列的OpenVaccine数据集,以及包含m6A修饰的mRNA序列的m6A修饰数据集。他们的结果表明,微调mRNA的降解和半衰期对于mRNA疫苗的安全性、有效性和正常功能是必要的。人类免疫学项目联合会(HIPC)(https://www.immuneprofiling.org),研究人员可以在特定感染、疫苗接种或佐剂治疗前后进行免疫系统的转录分析(包括microRNA阵列和下一代测序)。这将使对不同疫苗配方的效果和安全性进行全面评估,并加快人类疾病的评估。
通过创建用于组织收集、体外肿瘤培养和药物筛选的有价值工具,预计人工智能与类器官的整合将解决与传统预测相关的安全性和定制问题。通过集中关注抗原呈现途径和细胞坏死指数(CNI),人工智能也可以预测ICB反应。使用多重基因检测方法,Mizukami等人的系统生物学方法对疫苗安全性评估和在大鼠临床前调查中发现的某些指标,允许评估疫苗针对大流行H5N1流感的安全性。值得注意的是,目前有许多计算模型被用来预测基于可用数据集的蛋白质-蛋白质和药物-蛋白质相互作用。这表明人工智能模型有潜力模拟佐剂和疫苗之间的相互作用。这种能力不仅限于癌症疫苗,因为还有现有的佐剂数据库可以用于其他疫苗开发目的。例如,研究人员使用深度学习模拟了人类5′UTR序列与mRNA翻译之间的关联,提供了对潜在翻译效率的见解。CNN在识别DNA或mRNA序列中的基序方面表现出色,并在领域中比之前的非深度学习方法表现出优越性能,如基于卷积的架构,如基于深度学习的序列模型。然而,CNN在捕获远距离关系方面面临挑战,这对于涉及DNA和RNA的任务至关重要。像transformers这样的人工智能算法在预测DNA和RNA序列方面显示出前景。这些方法可以帮助优化疫苗开发并增强对分子过程的理解。领先的公司如Moderna在mRNA疫苗技术方面处于前沿,并在他们的研究过程中大量依赖人工智能。他们也正在开发三种癌症mRNA疫苗:mRNA-4157、mRNA-5671和mRNA-4359。此外,需要继续研究以提高人工智能引导预测的准确性,并微调准备这些疫苗所使用的技术。
5.2.肽疫苗
基于肽的癌症疫苗利用免疫系统启动针对肿瘤相关抗原的靶向反应,最终导致癌细胞的破坏。然而,在设计有效的癌症疫苗时面临的一个主要挑战是肿瘤内部异质性,这指的是单个肿瘤中癌细胞在遗传和表型特征上的重大变异性。这种变异性导致细胞群体的多样性,其中一些可能逃避免疫检测或对治疗产生抗性。免疫系统在癌症免疫监视中发挥着关键作用,通过识别和消除转化细胞来防止肿瘤形成。然而,以显著异质性为特征的肿瘤通常在各种亚克隆之间显示出复杂的相互作用,导致免疫细胞浸润和激活的波动。这种动态环境可能导致亚克隆对基于免疫的治疗反应不同,从而复杂化了免疫疗法方法的有效性。解决这一挑战需要一个多方面的策略,可以考虑到癌细胞的不同亚群。Stephens等人强调了精心设计基于肽的癌症疫苗的重要性,这些疫苗旨在特别针对并激活免疫系统的各个组成部分,特别是抗原呈递细胞。一个有前景的方法是基于新抗原的个性化肽疫苗的开发。这些疫苗旨在通过使用新抗原来增强免疫系统对抗肿瘤的能力,新抗原是每个患者癌症所特有的。由于人类群体中MHC等位基因的多样性,开发基于肽的疫苗是复杂的。用于预测T细胞表位的AI模型可能存在局限性,因为当抗原结合到细胞表面受体时,表位的空间配置会发生变化,导致潜在的假阳性和假阴性结果。此外,与MHC-I细胞相比,MHC-II限制性肽高度多样化且难以预测,因为它们的复杂性更高。生物信息学工具的进步,包括预测HLA等位基因覆盖率和使用能够结合多个MHC等位基因的多价肽,提供了解决肽疫苗设计的挑战的潜在解决方案。一个重大障碍是人与人之间MHC-I分子的巨大变异。很难预测哪些肽会附着在特定患者的MHC-I上,因为有数千种不同的MHC-I等位基因。免疫优势是另一个困难,因为并非所有肿瘤抗原都能同样有效地引发强大的免疫反应。肽的结构完整性也非常重要,因为它影响它们与T细胞受体的相互作用并启动有效的免疫反应。可能肽不足以触发强烈的免疫反应。为了解决这个问题,科学家正在研究使用佐剂,这些物质可以增强免疫系统并增加疫苗的效力。另一个必须解决的障碍涉及佐剂的创造和有效的递送方法,以确保肽有效地到达预期目的地。像SIMON(Sequential Iterative Modeling “OverNight”)这样的自动化ML系统比较不同临床数据集的结果,提高预测准确性并提供新的疫苗目标。像RF、SVM、递归特征选择、深度卷积神经网络(DCNN)、LSTM网络、NEC免疫档案器和免疫表位数据库(IEDB)这样的AI模型协助预测表位和设计有效的肽疫苗。ML也被用于设计基于肽的纳米材料用于肿瘤免疫疗法。此外,AI可以提供解决方案,以提高基于肽的抑制剂对免疫检查点阻断(ICB)的亲和力和稳定性,并使它们在临床环境中更有效。总之,基于肽的癌症疫苗有潜力成为对抗癌症的强大工具。然而,在抗原选择、佐剂使用和个性化方法方面仍存在挑战。像Ardigen这样的公司正在开发像ARDesign和ARDitox平台这样的AI工具来帮助设计基于肽的癌症疫苗。AI正成为解决肽疫苗设计挑战和提高其在癌症免疫疗法中效力的宝贵工具。Ardigen开发的ARDesign平台可以识别基于TCR的治疗方法的目标,并设计定制和现成的个性化癌症疫苗(PCV)。它包括以下模型:ARDitox、ARDisplay(展示模型)、Ardimmune(免疫原性模型)和Meta-model(考虑指标及其输出)。作为ARDesign平台的一个输入,DNA序列将与标准基因组进行比较,识别体细胞和生殖细胞突变。某些TCR与显著不同的表位序列之间的交叉反应已被报道。Sanecka-Duin等人开发了ARDitox,这是一种先进的计算免疫学和人工智能(AI)技术,用于检测和评估潜在的非目标结合。他们利用一组针对病毒表位的TCR和大量文献中的案例,其中TCR被用来针对与癌症相关的抗原,以评估ARDitox的计算效果。
5.3.树突细胞疫苗
DCVs是被激活的树突细胞,它们被暴露于癌症抗原中。它们被注入患者体内以引发对黑色素瘤细胞的免疫反应。DCVs中使用的成分包括从患者的肿瘤细胞中获得的抗原,在某些情况下,也可以使用如癌症干细胞等亚群。这些细胞的获取可以通过活检或手术切除肿瘤等方法实现。所选择的抗原通常是与正常细胞相比,在癌细胞中表达水平独特或升高的蛋白质。在实验室中,树突细胞随后被暴露于这些提取的抗原中。这种暴露过程可能涉及与肿瘤细胞裂解物或纯化抗原共同培养树突细胞。强调了从小规模试验到大规模试验中推断免疫反应的困难,以及对基于自体肿瘤裂解物策略而非共享抗原的偏好。DCVs针对肿瘤特异性肽或表位以诱导抗肿瘤效应。这些表位是来自TAA的短氨基酸序列,因其免疫原性和与HLA等位基因的兼容性而被选中。人工神经网络(ANNs)已被用于估计MHC分子与肽序列不同位置之间的结合强度。Mirsanei等人使用ANN数学模型来改进DCV递送。他们的研究旨在确定最佳的DC剂量和给药时间表,以增强基于DC的免疫疗法的有效性。ANNs可以处理纳米生物相互作用的数据,并预测最佳的DC疫苗接种参数,如DC类型、激活技术和注射部位。创建更优质的个性化DCVs涉及考虑各种因素,包括个体患者的生理信息、纳米颗粒特征和肿瘤变异。Paulis等人探索了基于DC的纳米疫苗的设计,用于肿瘤免疫疗法,强调了传递癌症抗原和免疫刺激信号以引发强大的抗肿瘤反应。Hashemi等人专注于使用纳米颗粒进行靶向药物传递至DCs,旨在提高基于DC的癌症免疫疗法中免疫反应的有效性和稳定性。纳米颗粒(NPs)对于促进强烈的T细胞和B细胞免疫反应至关重要,增加了细胞对免疫刺激代表的吸收,并经常作为自佐剂。尽管这些有利属性,对这些天然衍生的NPs与多种生物组分之间的相互作用进行全面探索,包括免疫细胞,仍然是必要的。这一彻底调查对于评估免疫毒性和推进免疫刺激NPs作为癌症免疫疗法中安全有效的工具至关重要。科学家们已经创建了能够预测为药物传递和癌症免疫疗法设计的纳米颗粒的理想尺寸、结构和表面属性的AI算法。例如,人工神经网络(ANN)被用来预测聚乳酸-羟基乙酸共聚物(PLGA)纳米颗粒的尺寸和初始释放速率。根据肿瘤模型、癌症类型和物理化学特性,NPs有所不同。癌症纳米医学的发展和研究可以从ML/AI与基于生理的药代动力学(PBPK)模型的整合中受益,正如Lin等人所展示的。他们的深度学习模型可以作为一个平台,帮助设计未来的癌症纳米医学,并帮助科学家决定哪些NPs应该进入临床前试验,从而减少并增强动物研究。在设计纳米结构和RNA纳米设计时,计算机辅助和数学建模可以帮助选择特定的构建模块以实现所需的结构。通过利用ML和AI模拟,纳米颗粒的设计可以促进对分子相互作用的理解。这种计算机辅助方法增强了对RNA和纳米颗粒相互作用的理解,从而增加了成功的可能性并优化了纳米颗粒的利用。由于与小分子、肽、受体、抗原和核酸的相互作用,特别是纳米颗粒在医学生物物理学中很有帮助。特别是,金纳米颗粒(Au NPs)在药物传递、癌症治疗和治疗诊断应用方面具有很大的前景。由于其最佳尺寸、形态和表面积,Au NPs是可适应的元素,提供了令人鼓舞的结果和支持。通过它们的完美细胞吸收和预防性细胞毒性措施,确保了最佳治疗条件。通过AI和数学建模的控制,Au NPs已经取得了进步,并将继续在医学生物物理学中发展。这篇专题综述强调了通过整合数学建模和人工智能(AI)来推进未来纳米技术研究的重要性,以增强医学生物物理学。Suberi等人开发了一种基于图像的技术,通过整合需要最少计算时间进行分析和调查的基于图像的技术,来增强疫苗制造。将AI整合到基于树突细胞的癌症疫苗的开发和优化中,有望提高其有效性和个性化。AI驱动的分析可以帮助确定最佳的肽序列、疫苗接种参数和疫苗制造过程,为更有效的癌症免疫疗法做出贡献。
6.佐剂开发与人工智能
佐剂是能够增强和/或塑造针对特定抗原的免疫反应的分子。像油包水混合物和铝盐这样的佐剂已经使用了多年。然而,许多佐剂在开发过程中面临挑战,例如稳定性、效力、耐受性或安全性问题。例如,铝盐可以诱导抗原聚集,从而影响疫苗的稳定性和免疫原性。基于角鲨烯的佐剂,如MF59和AS03,已与注射部位反应相关联。最近人工智能的进步已经通过计算机辅助分子设计和机器学习发现了两种新的广谱佐剂。尽管佐剂一直是疫苗接种的重要组成部分,但它们精确的作用机制以及它们对疫苗引发的免疫反应的增强或改变效果往往不清楚。本研究中使用的模型疫苗是一种自组装蛋白纳米颗粒(SAPN),展示疟疾环子孢子蛋白(CSP),并用三种不同的脂质体配方佐剂:脂质体加Alum(ALFA)、脂质体加QS21(ALFQ)和两者都有(ALFQA)。他们通过使用计算方法结合免疫分析数据来识别独特的疫苗诱导免疫反应。他们还构建了一个多变量模型,该模型能够仅基于免疫反应数据以92%的准确率预测佐剂条件。这作为一种有效手段来定位假定的免疫保护相关因素,这对于以逻辑方式匹配疫苗候选物和佐剂是必要的。相比之下,人工智能能够快速有效地分析大量分子和遗传数据,以找到可能增强免疫系统的潜在佐剂。此外,人工智能系统还可以模拟佐剂和免疫细胞之间的相互作用,使研究人员能够优化佐剂剂量和组合。这种优化导致产生理想的免疫反应,使得更有效和定制的癌症治疗的发展成为可能。佐剂在塑造疫苗诱导的免疫反应中发挥着至关重要的作用,通过各种复杂但通常微妙的机制,强调了它们对疫苗效力不可或缺的贡献。通过机器学习和深入分析疫苗诱导的细胞因子、细胞和抗体反应(称为“免疫分析”),可以识别出特定于佐剂的免疫反应特征。这些信息可以用来做出关于合理选择佐剂的明智决策。Chaudhury等人研究了由两种在临床上具有重要意义的相似佐剂,AS01B和AS02A,所引发的人类免疫反应的概况。他们识别了这些佐剂在疫苗诱导的免疫上留下的重要区分因素或免疫学特征。计算分析识别了一种可以以71%的准确率按佐剂分类参与者的免疫特征组合。此外,它还揭示了不同队列之间细胞和抗体反应的统计学显著变化。佐剂刺激对疫苗接种的强烈免疫反应。尽管已经使用了几十年,但今天使用的许多佐剂,如油包水乳液和铝盐,并不产生广泛或持久的免疫反应。因此需要更强的佐剂。通过利用机器学习和计算机辅助分子设计,Ma等人发现了两种新的广谱佐剂,有潜力增强疫苗反应。他们的库包括46种针对Toll样受体(TLR)的激动剂配体,这些配体在金纳米颗粒上合成。这项研究展示了计算机辅助设计和测试,可以快速识别有效的佐剂,以对抗当前疫苗接种相关的免疫下降。人工智能为加速癌症疫苗的佐剂开发提供了一个有希望的途径。它简化了有效佐剂的识别,优化了它们的使用,并增强了我们创建更有效和个性化癌症疗法的能力,同时节省了宝贵的时间和资源。
7.个性化癌症疫苗
个性化癌症疫苗是增强免疫疗法效力的一种有前途的策略,通过专门针对与癌症相关的抗原。在一项关键研究中,Ott等人证明,量身定制的新抗原疗法,结合检查点抑制剂抗PD-1,对晚期肿瘤患者,如非小细胞肺癌和膀胱癌患者,既耐受良好又有效。同样,在胶质瘤主动个性化疫苗联盟进行的I期GAPVAC-101研究中,Hilf等人将包含肿瘤特异性和共享抗原的高度个性化疫苗整合到标准疗法中。这种方法旨在最大限度地利用有限的目标空间,用于新诊断的胶质母细胞瘤患者。新抗原,源自特定于肿瘤的蛋白编码突变,在引发强烈的免疫反应中发挥着至关重要的作用。这些新抗原可以作为癌症疫苗的强大靶标,帮助排斥肿瘤。对于通常具有“冷”免疫微环境的肿瘤,如胶质母细胞瘤,使用多个表位的个性化新抗原疫苗策略已经显示出可行性。这种方法也在高风险黑色素瘤患者中进行了探索。Keskin等人进一步证明,来自外周血的新抗原特异性T细胞可以渗透到脑内胶质母细胞瘤肿瘤中,这是通过单细胞T细胞受体研究所揭示的。Sahin等人报告说,个性化的RNA突变体疫苗,无论是作为单独疗法还是与抗PD-L1结合,都触发了多特异性治疗性免疫反应,导致一些晚期肿瘤患者在临床上取得了客观的改善。这些发现强调了个性化癌症疫苗改善各种癌症患者结果的潜力。
人工智能驱动的进展将成为个性化肿瘤疫苗这一新兴领域的关键因素,标志着创新的前沿。开发定制的肿瘤特异性细胞疫苗(TCVs)的一个关键的初始步骤是识别存在于癌细胞外部的肿瘤特异性新抗原(TSNs)。将人工智能整合到这一过程中显著加速了对潜在癌症疫苗的识别和选择,以适应个别患者的需求。通过利用机器学习(ML)将体细胞突变转化为可操作的新抗原,人工智能提供了更有效的免疫反应选择,这些反应具有最大的治疗潜力。此外,其他计算方法,如将遗传算法与支持向量机(SVMs)相结合,已在疫苗开发中实现了高预测准确性。其中一个工具,PREDIVAC,在识别CD4+ T细胞表位方面表现出色,超越了现有的预测HLA II类肽结合的方法。将计算分析、高通量基因组学和机器学习整合起来,可以显著简化治疗靶标和反应生物标志物的识别,这对于开发有效的个性化癌症疫苗至关重要。然而,要在这一领域取得成功,将需要开发可解释的人工智能系统,能够解释其结论背后的逻辑,确保对人工智能辅助决策过程的透明度和信任。
8.使用人工智能开发癌症疫苗面临的开放性挑战
强调了在训练人工智能模型时大型、高质量数据集的关键重要性,需要准确和相关的信息。为了开发能够处理噪声数据并泛化到未经训练样本的强大模型,至关重要的是要确保输入这些模型的数据既全面又精确。这在预测B细胞表位时尤其重要,它们在蛋白质中的位置、大小和序列差异很大。预测抗原表位的挑战在于识别能够与抗体结合的蛋白质区域,当考虑整个蛋白质复合物时,这项任务变得更加复杂。在多链蛋白质结构中预测抗原表位需要模拟链之间的相互作用,与单链蛋白质预测相比增加了复杂性。此外,抗原表位受到宿主免疫的选择压力,使它们比标准结合位点更具变异性。这种变异性使得抗原表位预测不适合传统的结合位点预测技术。相比之下,一些采用机器学习技术的构象B细胞表位预测算法,结合了保守性、结构、几何和氨基酸属性等特征,在准确性方面优于结合位点预测模型。然而,即使有了这些进步,克隆多样性和原发性及转移性肿瘤内部的肿瘤异质性对依赖突变的新抗原预测提出了重大挑战。并非所有新抗原都能作为癌症疫苗的有效靶标,这突出了识别免疫相关新肽的困难。对于突变负担较低的癌症,对不依赖突变的新抗原的需求变得更加关键。不幸的是,大多数当前的人工智能模型主要关注依赖突变的新抗原预测,常常忽视了肿瘤可及性等重要因素。新抗原预测的一个关键方面是HLA等位基因的作用,它们向免疫系统呈现抗原。准确的新抗原预测必须考虑肽突变与患者独特HLA等位基因的对齐。通过纳入这种关系,研究人员可以更有效地识别能够引发强大免疫反应的新抗原,这对于开发个性化癌症免疫疗法至关重要。将这些特征整合到预测算法中有可能显著提高其性能。肿瘤微环境中的T细胞迁移可能会遇到障碍,包括细胞外基质和与癌症相关的巨噬细胞,这可能会限制T细胞接触肿瘤抗原。肿瘤可及性,受血管化不良、物理屏障和限制T细胞渗透的血管存在等因素的影响,是新抗原特异性T细胞反应产生后的一个关键阶段,但由于数据集限制,这一方面在当前预测工具中经常被忽视。单细胞分析可以用来解决批量测序在捕捉肿瘤免疫微环境异质性方面的不足。疫苗效率还受到T细胞无法检测免疫逃避肿瘤和T细胞被免疫抑制性肿瘤微环境(TME)抑制的影响。已经有人尝试对TME进行建模,但当前的人工智能模型在检测免疫逃避肿瘤方面存在困难。个性化新抗原癌症疫苗还面临T细胞耗竭和功能障碍的挑战,其特征是效应功能丧失、增强的抑制受体表达和细胞死亡倾向,这显著限制了它们的好处。对于推进下一代癌症疫苗而言,早期预测疫苗反应的签名至关重要,需要持续进行研究。
9.未来展望
癌症疫苗设计的未来研究应优先考虑旨在增强免疫细胞招募、增加表位表达,并选择能够激发针对肿瘤相关抗原的强烈T细胞反应的最佳表位组合的策略。分子测序、人工智能(AI)和细胞工程的进步有潜力彻底改变癌症疫苗,使它们更快、更经济、更有效。这些技术允许对癌症疫苗接种的免疫反应进行快速和全面的评估,促进基于个体患者反应的实时调整。数字化转型的临床研究在预测不同条件下的患者反应中发挥着关键作用,导致更精确的疫苗开发。机器学习(ML)和随机优化技术的应用有助于识别最佳疫苗接种策略,确保以最小剂量实现效力。AI技术可以预测黑色素瘤细胞的潜在变化,使疫苗能够保持对驱动突变的有效性。此外,AI可以与VARE(疫苗不良事件报告系统)等系统合作,识别可能面临潜在疫苗接种风险的人群。除了预测建模外,AI模型和合成生物学的发展导致了生物机器人和异种机器人的创造,它们在癌症治疗中有潜在应用。这些创新实体可能在药物输送到肿瘤靶向等方面发挥作用,扩大了癌症治疗的可能性。AI、分子进步和生物机器人之间的协同作用代表了一种多方面的方法来推进癌症疫苗设计和交付。AI促进基于个体患者反应的实时调整、预测建模和识别最佳疫苗接种策略。然而,数据隐私、算法偏见、监管合规(203)和公平获取等伦理和法律考虑至关重要。纳米计算机是控制或指导体内纳米机器人的设备。它们可以是电气的、生物的、有机的或量子的。使用DNA中的四个氮碱基字母编程的软件可以控制由DNA在分子水平上构建的计算机中的基因表达。它还可以识别与特定基因相关的mRNA类型,这些基因在过表达或相反地低表达时,可能有助于癌症的发展。这使得诊断各种癌症形式并用推荐的药物治疗疾病成为可能。2016年,研究人员开发了旨在针对并摧毁脑癌细胞的模拟纳米机器人。这些纳米机器人具有识别和根除癌细胞的能力。在检测到肿瘤后,它们发出听觉信号,便于后续干预的精确定位。人工智能(AI)的出现为社会带来了许多法律和伦理困境,包括与隐私和监控、偏见和歧视以及人类判断的作用相关的担忧。这些挑战也可能提出哲学复杂性。新型数字技术的出现引发了它们可能引入额外的误差和数据泄露来源的担忧。在医疗领域,流程或协议中的错误可能对成为这些错误的受害者的患者造成灾难性后果。寻找伦理和法律问题也可能是减少风险的积极方式,这将有助于AI技术的更广泛使用和总体成功。通过解决伦理问题和保证监管合规,可以促进AI技术应用的透明度,这可以提高患者信任——这是医疗保健的一个关键组成部分。评估免疫疗法有效性的另一个潜在方面是AI识别淋巴细胞、肿瘤细胞和间充质基质切片的能力,并使用3-D建模强调各种细胞类型的空间分布。对于免疫疗法,一个最佳的基于AI的预测模型应该包含所有相关的临床数据,如遗传学、影像学、蛋白质组学、病理组织、人口统计数据、病史等。为了允许从许多地点共享大量数据,加强数据收集的完整性和客观性至关重要,因为像泛癌分析这样的概念已经在评估PD-1/PD-L1效力中得到反映。这对免疫疗法的未来非常有利。临床医生越来越关注深度学习(DL)神经网络,因为它们在跟踪和预测治疗反应方面表现出良好的性能。如今,实体瘤诊断(胃肠道肿瘤、肺癌、黑色素瘤等)是使用生物标志物分析、计算机化组织病理图像解释或自动量化放射成像完成的。然而,关于使用AI评估非小细胞肺癌(NSCLC)免疫疗法效力的研究并不多。Russo等人的计算框架有潜力加快和改进设计和开发最佳疫苗配方的过程,这可以增强免疫系统的反应,从而提高治疗性疫苗的效率并保护预防性疫苗。使用in silico试验技术,这个计算管道的优势可以加快和改进为最有希望的疫苗候选者设计第I/II阶段临床试验。IST正在做很多工作以获得监管机构的合格印章。目前,医疗环境缺乏明确的立法来解决使用人工智能可能引起的伦理和法律问题。在创新和伦理保障之间取得平衡对于确保负责任地开发和部署AI驱动的癌症疫苗至关重要。
9.1.总结
癌症疫苗研究集中在通过增强招募和表位选择来改善免疫反应。分子测序、人工智能(AI)和细胞工程等先进技术有望实现更快、更有效的疫苗开发。AI能够根据个体患者反应进行实时调整,而纳米计算机和纳米机器人提供了创新的药物输送解决方案。然而,必须解决包括数据隐私和算法偏见在内的伦理和法律考虑,以确保负责任地部署AI驱动的癌症疫苗。在创新与伦理保障之间取得平衡对于安全有效的癌症治疗至关重要。
10.讨论
尽管试图汇编多样化的研究,但综述承认由于数据可用性主要在英语语言中,存在局限性。这种语言限制提高了对可能排除非英语文献中宝贵见解的认识。这强调了未来综述采用更全面的语言方法的重要性。采用PRISMA-ScR方法论来减少数据收集中的个人偏见。此外,声明综述论文不对引用的研究报告的结果负责,这引发了对包括研究的可靠性和有效性的质疑。这促使人们考虑基础研究的方法论严谨性及其对综述发现的整体稳健性的影响。此外,故意强调来自知名期刊的高质量论文可能会引入潜在的偏见,在解释综述的结论时应承认这一点。这强调了对排除知名度较低期刊研究的局限性有细微理解的重要性。综述通过强调开发有效癌症疫苗的复杂挑战,包括目标识别、免疫耐受和患者特定变异等方面,做出了重大贡献,指导研究人员穿越这一复杂领域。综述还在解释癌症疫苗设计中应用的基本生物学概念和AI工具方面发挥了关键作用。研究探讨了生物标志物预测、表位设计、MHC结合预测和免疫原性预测,将传统生物学与先进的AI应用相结合。详细探讨各种癌症疫苗类型,包括DNA、mRNA、肽和树突细胞疫苗,增强了理解。包括总结数据集、疫苗开发研究中的AI技术以及正在进行的临床试验的表格,为研究人员提供了宝贵的资源,促进了数据收集和比较分析。这些表格还提供了当前研究格局的见解,并指导未来的研究工作。除了其具体贡献外,综述还启动了关于阻碍癌症疫苗广泛采用的挑战的更广泛讨论。它深入探讨了其有限使用的原因,检查了临床采用和监管方面的挑战,为治疗性癌症疫苗开发的现状提供了背景。综述还将重点扩展到疫苗设计之外,探索AI在设计临床试验、寻找潜在疫苗接种风险人群和选择最佳剂量方面的潜力。这种更广阔的视角强调了AI的变革性影响,不仅在疫苗开发中,还在重塑癌症治疗的不同方面,包括患者分层和个性化治疗策略。
11.结论
癌症疫苗的出现代表了全球癌症治疗的重要进展。在疫苗开发过程中利用基于AI和ML的技术,能够精确预测和识别能够触发强大抗肿瘤免疫反应的新抗原。癌症疫苗发现的最新进展部分解决了与抗原选择相关的挑战。这些新方法包括多种机制,以抵消癌细胞的免疫抑制效应,为个性化癌症疫苗开发铺平了道路。AI在定制这些疫苗方面发挥了关键作用,为传统疗法的局限性提供了有希望的解决方案。为了获得最佳结果,在疾病进展的早期或最小残留阶段部署癌症疫苗似乎是最有效的。相关领域如人工智能、细胞技术和DNA测序的进步可能使这些疫苗受益,所有这些都有助于检查和优化疫苗诱导的免疫反应。虽然目前用于设计核酸疫苗的计算技术,如表位预测和序列优化,由于技术发展,目前表现出适度的性能水平,但广泛的在线数据和先进模型的可用性带来了巨大的希望。ML算法可以预测表位,并已整合到几种预测方法中,尽管存在一些准确性限制。其中一个困难是用于开发和评估临床环境中AI模型的高质量数据供应有限。尽管通过GAN和扩散模型创建合成免疫原性肽模型相对未被探索,但这些工具已在计算机视觉和合成生物学等领域取得成功,生成了感兴趣的新图像和序列。AI为个体癌症患者量身定制治疗方案并加速开发新技术的潜力,有望彻底改变肿瘤治疗领域。免疫组学有望在未来的肿瘤免疫学中发挥领导作用,但仍有大量工作要做。特别是在免疫基因组学和单细胞分析方面,AI的进步预计将显著推进该领域的临床应用。随着实验验证数据的更大可访问性和改进算法的持续开发,由广泛数据集驱动,将增强疫苗设计策略。虽然AI为加速癌症疫苗设计提供了一个有希望的途径,但必须承认其局限性,特别是关于生物系统的复杂性和临床转化的挑战。尽管AI驱动的模型具有预测能力,但它们通常受到数据可用性和质量的限制,它们的预测可能无法完全考虑到免疫系统和肿瘤微环境的动态性质。免疫逃避、肿瘤异质性、免疫反应的不可预测性以及个体遗传变异等生物学因素可能显著影响疫苗在临床环境中的有效性。因此,应将AI视为与传统实验方法一起工作的辅助工具,而不是单独的解决方案。实验验证、动物模型和严格的临床试验仍然至关重要。总之,AI驱动的癌症疫苗设计和开发在学术界和工业界正受到越来越多的关注。各种公司正在向该领域投入大量资源,以解决当前挑战,提供使用AI的定制解决方案。治疗性癌症疫苗的关键作用被肿瘤生物学和疫苗技术的发展所突出,特别是在早期或最小残留疾病的情况下。肿瘤疫苗的发展进步是由对有效癌症治疗的迫切需求所驱动的。这些进步可以从包括免疫学和癌症生物学在内的多学科合作中以及人工智能(AI)的整合中大大受益。
识别微信二维码,添加生物制品圈小编,符合条件者即可加入
生物制品微信群!
请注明:姓名+研究方向!
版
权
声
明
本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(cbplib@163.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。
疫苗免疫疗法信使RNA
100 项与 Scopus Biopharma, Inc. 相关的药物交易
登录后查看更多信息
100 项与 Scopus Biopharma, Inc. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年11月05日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
药物发现
2
7
临床前
登录后查看更多信息
当前项目
| 药物(靶点) | 适应症 | 全球最高研发状态 |
|---|---|---|
DUET-201 ( TLR9 ) | 实体瘤 更多 | 临床前 |
DUET-102 ( STAT3 x TLR9 ) | 胶质瘤 更多 | 临床前 |
DUET-02 ( STAT3 x TLR9 ) | 前列腺癌 更多 | 临床前 |
DUET-03 ( STAT3 x TLR9 ) | 肿瘤 更多 | 临床前 |
WINi(Vanderbilt University) ( WDR5 ) | 其他血液系统恶性肿瘤 更多 | 临床前 |
登录后查看更多信息
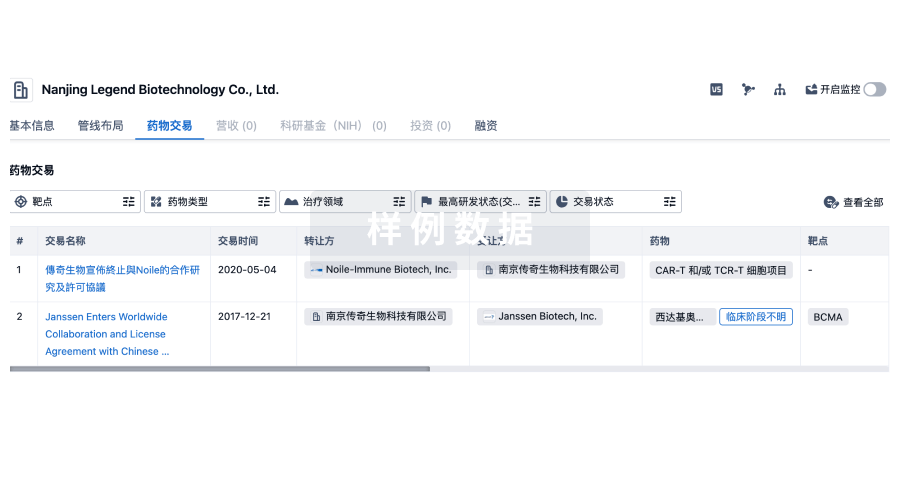
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
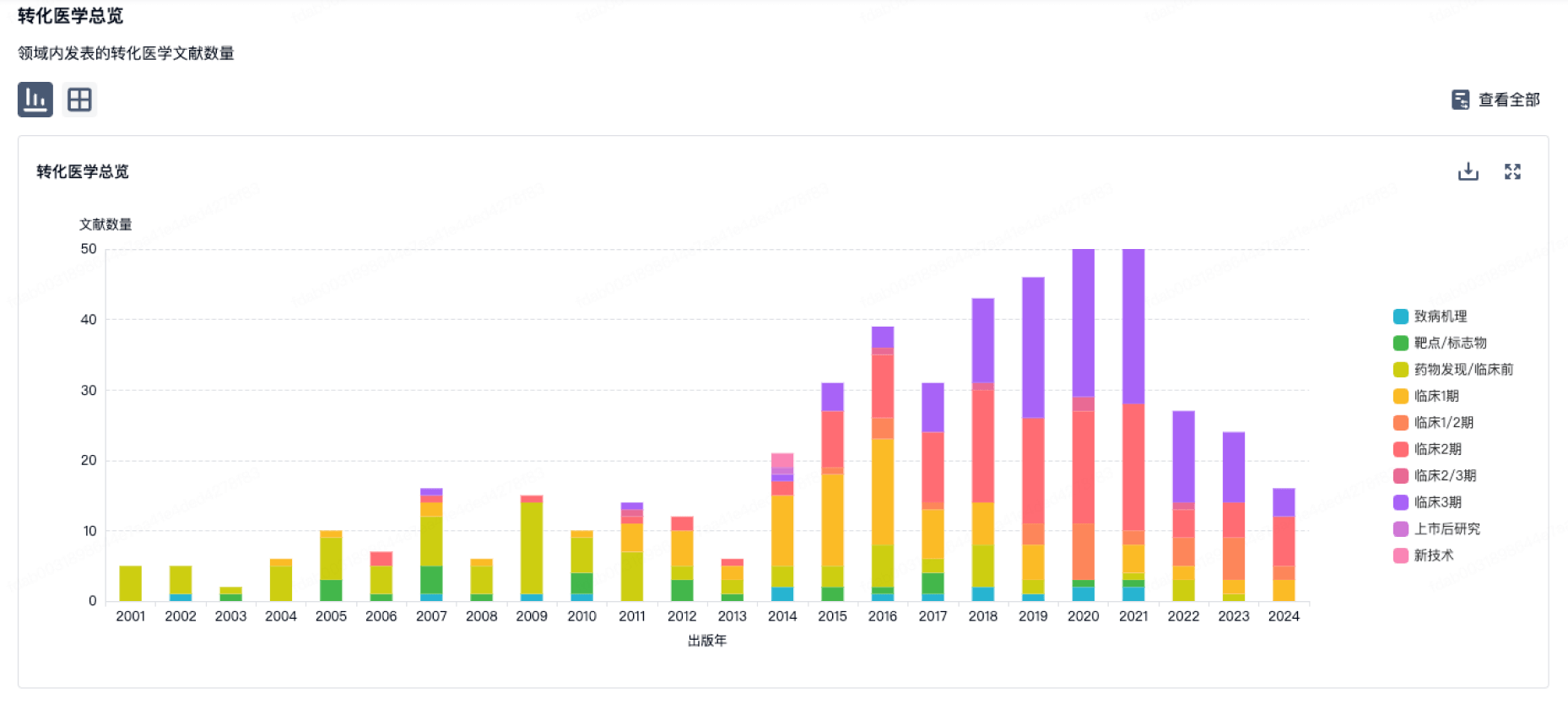
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
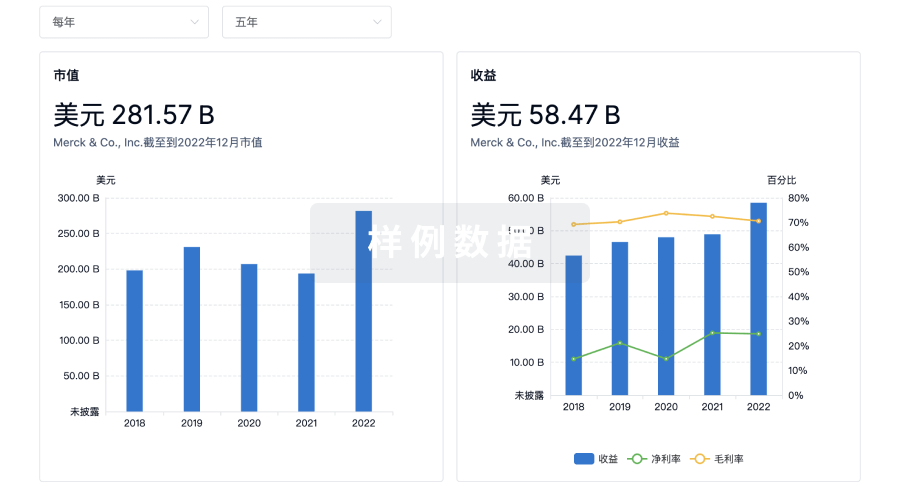
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
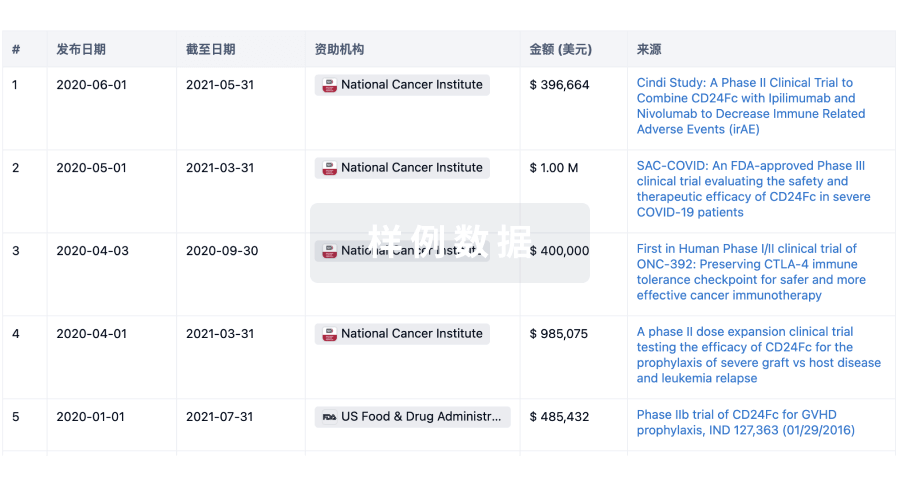
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用