更新于:2024-05-01
Hassab Labs
更新于:2024-05-01
概览
关联
1
项与 Hassab Labs 相关的临床试验Four Year Outcome After Severe Traumatic Brain Injury in the Parisian Area
The primary objective of the protocol is to study the long-term outcome of a large group of traumatic brain injury patients.
This outcome is to be described in terms of activity, participation, quality of life, SOCIO-professional outcome and impact on caregivers, and in relation to health care provision.
The secondary outcome is to measure the impact on functional outcome of several predictive factors, and their relative importance on outcome. Our principal hypothesis is that SOCIO-professional and health provision factors play a major role on long-term outcome, further even than initial severity of brain injury.
This outcome is to be described in terms of activity, participation, quality of life, SOCIO-professional outcome and impact on caregivers, and in relation to health care provision.
The secondary outcome is to measure the impact on functional outcome of several predictive factors, and their relative importance on outcome. Our principal hypothesis is that SOCIO-professional and health provision factors play a major role on long-term outcome, further even than initial severity of brain injury.
开始日期2010-02-01 |
申办/合作机构 |
100 项与 Hassab Labs 相关的临床结果
登录后查看更多信息
0 项与 Hassab Labs 相关的专利(医药)
登录后查看更多信息
3
项与 Hassab Labs 相关的新闻(医药)2024-02-27
·药研
摘 要:孤儿药的定价和报销关系到患者用药可及,对罕见病的防治与保障具有重要意义。欧洲国家在孤儿药医保准入过程中形成了卫生技术评估的特殊标准和路径,并建立了专项支付基金和多样化的风险分担协议,有效提升了孤儿药的可及性。基于此,文章选取欧洲典型国家,对比分析其孤儿药定价和报销的主要特点和共性保障思路,提出“构建孤儿药卫生技术评估加速程序、探索医保外孤儿药的分类保障机制、通过孤儿药定价和调整实现供给激励和研发激励”的建议,以期为优化我国孤儿药市场准入机制提供参考依据。孤儿药具有目标市场有限、研发成本高的特点[1],企业研发动力不足,仅靠市场机制调节并不能满足患者的用药需求,需要政府利用价格干预、定价激励等方式,保障孤儿药的可获得性。另一方面,价格高昂的孤儿药如果不纳入报销范围,将给患者带来较大的治疗负担;但纳入报销又将给医保基金带来较大的冲击,转化为重大的预算影响[2]。由此,从平衡孤儿药定价激励和医保控费的视角出发,优化孤儿药的定价和报销机制对提高我国罕见病患者用药可及意义重大。在欧盟,孤儿药虽通过EMA 集中程序注册上市,但“定价和报销”是各成员国的责任[3]。因此,欧洲各国在孤儿药的定价和报销中实践经验丰富,对我国具有一定的借鉴意义。欧洲国家大都已建立特殊的卫生技术评估(health technolog yassessment,HTA)标准,并形成了高值孤儿药报销的特别方案;荷兰、比利时、奥地利和爱尔兰等地还建立了“BeNeLuxA(Ir)”联盟,通过互认HTA 结果,以联盟谈判的方式进行孤儿药的合作定价和采购。同时,部分国家通过多样化的风险分担协议,平衡药品风险和早期获益,缩短了患者获取药物的等待时间。1 欧洲国家孤儿药的定价和医保准入机制1.1 英国的定价和医保准入机制英国先由制药公司自由定价,之后政府将通过药品价格监管计划和NICE 卫生评估的结果对价格进行调整,形成最终定价[4]。新药准入则是在接受HTA 之后,经NICE 推荐纳入到英国国民健康保障体系(Nationa Health Service,NHS)获得全额报销[5]。药品进口注册流程、申报资料撰写与上市后维护高级研讨班在 HTA 过程中,针对孤儿药,NICE 往往会接受更低的证据水平和更高的增量成本效益比(Incrementally Cost-Effectiveness Ratio,ICER)阈值[6]。对于ICER仍然超出阈值的孤儿药,NICE 将从孤儿药的社会收益、创新性、疾病患病人数、疾病的特殊性等多方面进行综合考虑,提高孤儿药纳入报销的可能性。此外,英国针对治疗超罕见疾病(患病率低于1/50 000)的孤儿药建立了高度专业化技术评估方案,其评估委员会由国家医疗服务体系人员、学术界、制药和医疗器械行业代表,患者代表、其他卫生专业人员等构成,引入了更多的价值评估维度。1.2 法国的定价和医保准入机制法国由国家卫生委员会(Haute Autorité de la Santé,HAS)下设的透明委员会(Transparency Committee, TC)承担新药的卫生技术评估工作,主要评估实际临床效益(Service Médical Rendu,SMR)和附加临床效益(Amélioration du ServiceMédical Rendu,ASMR),并分别决定报销比例和定价方式(如图1)。图1 法国孤儿药的卫生技术评估及定价报销程序从评价结果看,根据欧盟有关罕见病的认定标准,罕见病属于严重疾病[7],所以孤儿药的SMR 评估结果都较为积极,且不再要求企业提交经济证据,报销比例多在35%以上[8]。同时,由于罕见病目标适应症下治疗方案较少,孤儿药均能够获得ASMRI~III 级评价,不必受到量价协议和同类药品价格的限制,获得较高的定价水平。HAS 还特别规定,如果孤儿药目标适应症的预算影响小于每年3 000 万欧元,则直接获得ASMR I 级评级。从评估过程看,HAT 明确孤儿药根据疾病严重程度和临床急需性,可以豁免成本效益评估。且对属于创新先驱(priori innovative)的孤儿药,还可以通过加速HTA 程序,在新药上市前更早地开始HTA 评估,提前收集卫生技术评估的相关信息,从而加快HAS 的决策过程。1.3 荷兰的定价和医保准入机制荷兰主要通过“外部参考定价”的方式进行定价,即参考欧盟其他成员国的价格确定最终定价。此外,荷兰还通过“BeNeLuxA(Ir)”联盟进行谈判定价[9]。联盟主要通过互认卫生技术评估结果,提高药品定价和报销的效率,以更为透明的机制进行合作定价;通过联合价格谈判和市场调查,提高支付方在市场上的地位。对于孤儿药,BeNeLuxA(Ir)正在探索以几个国家联合谈判的方式“团购”孤儿药,并进一步降低药价。在HTA 过程中,荷兰纳入了多个利益相关者的评价维度,且十分关注新药的社会性。对于孤儿药,由于其具有更加明显的社会价值,荷兰依据医疗保险委员会(CVZ)会直接免除成本效益评估,并依据附加效益、疾病罕见性、严重程度等指标在给予其更为积极的HTA 结果。在医保准入上,CVZ 提供的HTA 报告和报销建议形成最终的报销决定。由于孤儿药往往具有临床比较优势,多数都能够获得全额报销。对于部分临床价值证据不完整、成本效益不确切或预算影响不满足要求的孤儿药,也可以申请纳入高值/特殊报销清单获得早期报销。但是,列入这类清单的药物需要提交预后成本预测和研究计划,在报销期限内持续收集临床证据,以方便后续评估[10]。1.4 欧洲国家孤儿药定价和医保准入的特点总结1.4.1 欧洲孤儿药定价和报销的特点。(1)孤儿药定价和报销依赖HTA 结果英国、法国、荷兰都通过HTA 来为定价和报销提供决策依据,并设立有专门的评估机构和专门的评估标准。HTA 的评级将在很大程度上影响孤儿药的价格水平和定价方式,并决定实际报销比例。(2)给予孤儿药科学的溢价空间。虽然每个国家HTA 立足的方法基础和价值确定方式不同,但均关注孤儿药相较现有疗法(产品)的附加效益和临床必需性。所以经过欧盟认定的孤儿药均能够获得积极的评价,从而能够以外部参考价的定价方式进入市场,给予了孤儿药很大的溢价空间。(3)以参考定价为主,探索联盟采购机制。在定价过程中,欧洲国家多采取“参考定价”和“谈判定价”相结合的方式,以“参考定价”确定药品的限价基准,然后结合HTA 评价结果和制药企业的自主报价来进行协商,确定最终药价(表1)。药品注册经理能力全面提升与实践案例分享(注册必备)对于荷兰、比利时等议价能力有限的国家,已经在逐步探索通过借助联合价格谈判和市场调查的方式, 提升支付方的议价地位,呈现“团购”趋势。1.4.2 欧洲国家孤儿药HTA 的特点。(1)建立孤儿药的特殊评估标准或路径。在评估标准上,由于孤儿药价格高昂且临床数据不足[11],各国都针对孤儿药建立了特殊的评估标准,包括提高增量成本效益阈值、增加多维度评价标准(如纳入患者及家属视角)、降低临床数据要求(如法国在效益评级过程中直接认可孤儿药的附加效益,简化数据提交)等。此外,部分国家还形成了特殊的卫生技术评估路径,如英国采取高度专业化技术评估方式评估超罕见疾病用药;法国对孤儿药设置了加速HTA 程序,在上市前即开展评估,加速孤儿药上市后的市场准入。(2)联盟采购关注HTA 的透明度和清晰度。BeNeLuxA 联盟的目标是通过合作HTA,达成联合定价和报销。目前BeNeLuxA 联盟主要确定了四种合作方式(表2)。这些合作方式均建立在统一的 HTA 标准之上,对孤儿药HTA 评价的透明度和清晰度有较高的要求[12]。而合作谈判既节省审评资源,又提高高值孤儿药的可获得性,具有一定的实践意义。2018 年,比利时将荷兰对诺西那生钠注射液的HTA 报告用于报销决策中,并通过联合谈判降价纳入到了BeNeLuxA 联合采购系统当中,实现了高值孤儿药的降价准入[13]。(3)降低孤儿药HTA 中成本效益分析要求。从评估内容看,各国均涉及附加效益评估和预算影响分析,但对成本效益的分析要求有所降低(表3)。由于各国罕见病自然史研究薄弱,孤儿药在上市后的疗效评价、成本效益分析和增量成本效益计算中都较为困难,早期成本-效益数据明显不足。因此,在评估中,基于疾病严重程度、临床急需性等标准,法国和荷兰在评估中豁免了成本-效益分析的要求。2 欧洲国家孤儿药的特殊报销策略2.1 英国孤儿药的特殊报销策略对于被NICE 拒绝推荐或者仍然处于评估状态的孤儿药,英国设立有特殊的资助项目进行补充保障,如个人基金请求、肿瘤药物基金、专项购买服务和对缺乏临床数据项目的评估购买计划。同时,英国建立了患者准入计划(patient accessschemes,PAS)和管理准入协议(Managed access agreements,MAAs)进行孤儿药的支付。PAS 是孤儿药最常用的创新支付方式,旨在提高成本效益,使患者能够获得高值孤儿药[14,15]。MAAs 则是一种有条件报销的协议。协议签订时,NHS 将设定具体的报销条件(如覆盖人群、结局指标和折扣价格等)、协议持续时间、数据收集方法和报告频率。期间企业将以商定的折扣价格提供药品,并持续收集真实世界的数据,以加强药物的证据体系,获得长期报销。2.2 法国孤儿药的特殊报销策略对于基本医保覆盖之外的孤儿药,法国还采取了两项特殊报销策略。其一是长期疾病清单(Affection Longue Durée,ALD),因严重程度或慢性性质,需要长期护理和高成本治疗的疾病将被纳入清单中,获得全额报销[16]。罕见病多为慢性病、严重疾病,往往可以被列入ALD 当中,对应的孤儿药也可获得全额报销。其二,法国建立了临时使用授权(Authorisations for Temporary Use,ATU)计划,相当于同情用药制度,旨在为罕见病患者提供尚处于临床试验阶段的孤儿药。在ATU 计划内的孤儿药批准上市后,将以post-ATU 的方式直接获得报销,直到最终决定纳入报销范围并确定最终价格,缩短了孤儿药纳入报销的时间[17]。2.3 荷兰孤儿药的特殊报销策略对于未纳入报销的高值孤儿药,荷兰引入了基于证据发展的报销(coveragewith evidence development,CED)协议[18]。政府通过与制药企业签订协议,降低对孤儿药早期临床数据和关键证据的要求,使得孤儿药可以更早得到偿付。企业则需要在协议期内,持续开展研究和证据收集工作,以验证药品带来的健康产出。协议到期后,政府根据对上市后数据收集结果确定其健康效益,而后形成最终报销决策。2.4 欧洲国家报销策略的特点总结2.4.1 重视发挥风险分担协议的作用。为保障罕见病患者用药的可及性,欧洲国家通过签订多样化的风险分担协议(Risk-Sharing Agreements,RSA)的方式,有效缩短了患者获得药品等待时间(表4)。总体来看,3 个国家涉及的风险分担协议主要有两种类型,一种是基于财务预算的管理协议,比如英国的MAAs、法国的post-ATU 报销。该类协议一般会设定成本上限或数量上限,在限定时间内规定每位患者的最大累计治疗成本或治疗计量,超过此阈值,药品制造商以折扣价格或免费提供其药物,也可能需要向支付方退款。此类协议容易操作,且管理的要求较低,应用更为广泛[19];另一种是基于健康产出结果的风险分担协议,比如英国的PAS 和荷兰的CED。这类协议则主要关注药物预期的临床效果,在某个时间节点或健康结局如果达不到承诺的效果指标,制药企业需要向支付方退款。2.4.2 构建高值孤儿药特殊支付清单。欧洲国家通过设立特殊的报销清单的方式(表5),为没有纳入医保报销范围的高值孤儿药开辟了新的报销路径。这类清单或专项基金的筛选标准可能是以“疾病”为依据,关注疾病的性质(难治性、慢性)、疾病严重程度、治疗成本等等;也可能以“药品”为依据,关注药品当前的疗效证据、定价潜力、临床急需性等等。2.4.3 关注孤儿药上市后数据收集。无论是孤儿药的管理准入协议还是有条件报销路径,都采用了“临时报销” 的共同思路,要求企业在上市后持续收集临床使用证据和真实世界数据。并通过再评估门槛的设置,提升了企业数据收集和分析的主动性。3 启示和建议3.1 构建孤儿药准入评估体系,建立加速HTA 程序建议在医保准入过程中,构建孤儿药配套的卫生技术评估体系。一方面,孤儿药临床试验招募困难,要构建完整的数据链也存在客观障碍。所以,应在评估中降低对部分数据的要求,更关注其附加临床价值。另一方面,罕见病作为一个社会性问题,应将社会效益、公平性原则等标准纳入考虑之中,减少对成本-效益分析的依赖性,构建多维度的评估体系。同时,建议我国针对孤儿药建立加速 HTA 程序。构建评估专家团队早期介入到III期临床试验中,在孤儿药上市前按照HTA 要求收集评估所需要的数据及材料,加速孤儿药在上市后的医保准入。3.2 探索孤儿药“分类”保障机制,提升药品可及为平衡医保基金影响及患者用药需求,对于尚不满足医保纳入条件的孤儿药,建议分类进行支付保障。3.2.1 建立高值孤儿药报销清单,拓宽资金筹措渠道。对于高值孤儿药建议成立专项支付基金予以报销,通过财政拨款、社会捐赠等多种方式募集基金,发挥多层次医疗保障的协同支持作用。从管理上,我国目前尚未形成孤儿药认定程序,故可针对高值的孤儿药采取“清单”管理的方式。通过设立一定的纳入标准,如疾病严重程度、替代疗法情况、临床急需程度等等。优先保障罕见病目录中纳入、且无替代治疗方法的创新孤儿药,做好罕见病目录与孤儿药报销清单的有效衔接。3.2.2 丰富风险分担协议类型,促进上市后数据收集。对于临床证据不足的孤儿药可引入风险分担协议,对上市前期的风险进行控制。建议在我国在前期探索经验的基础上,对于早期临床效果数据不充分的创新孤儿药,引入基于临床效果指标的风险分担协议,以促进长期的临床证据收集和罕见病自然病史的研究。并不断丰富风险分担协议类型,规范协议的制定、执行及管理流程,扩展应用范围。3.3 巧用溢价激励,配合价格调整机制中国作为亚洲国家定价的参考国之一,过低的溢价比例将会影响制药厂商进入中国市场和保障供应的积极性。因此,建议我国在谈判和采购过程中,选取合理的采购方式,给予孤儿药一定溢价比例,激励本土研发和引进。对于一些不具有临床替代性的廉价孤儿药,因利润有限出现短缺的,也可以采取反向的调节机制,允许孤儿药根据临床疗效评价结果和该年的销售情况获得一定的成本补偿。4 结语罕见病作为一个国际性的公共卫生问题,解决好孤儿药的可及问题具有较高的社会价值。因此,为鼓励罕见病领域的药品创新,我国仍然需要保持溢价激励和价格调节机制,但为平衡医保控费,保障药品可及,还需进一步优化孤儿药的评估和支付方式。在卫生技术评估中应充分体现孤儿药的临床价值及社会价值,做好孤儿药上市后疗效数据和安全性数据的收集。在孤儿药的支付过程中,有效发挥风险分担协议、高值孤儿药基金的补充支付作用,优化多层次医疗保障体系。------------THE END------------内容来源:《中国卫生经济》点击查看:投稿获丰厚稿费关注药研 一路同行药研论坛:始终以为药品研发一线人员提供高质量、高性价比培训为第一宗旨。自成立以来,累计举办80余期药品研发、注册领域研讨班。据统计,中国医药工业百强企业和研发百强企业均超过90%参加过药研收费类培训。截至2023年底,包括上海强生、辉瑞、阿斯利康、山德士、日本大冢、大鹏、卫材、小林制药、扬子江、恒瑞、正大天晴、东阳光、科伦、中科院等在内的2800余家企事业单位参加过药研线下收费培训,得到业界普遍认可与好评!药研学院:已同众多企业合作超百期直播课,全网观看突破100万+,药研直播课聚焦药品研发相关主题,平均观看人数行业领先。药研自媒体矩阵20万+:目前药研公众号研发领域关注用户约10万人!双直播平台10万人,微信社群5万人,其中制药企业和研发机构关注量5000+。商务合作:15911172616
孤儿药
2022-11-13
2022 年 1 月 11 日,欧盟卫生技术评估条例(Regulation (EU) 2021/2282)在经过多年的谈判后生效。欧盟在卫生技术评估方面的合作旨在帮助欧洲的患者获得创新的健康技术,更好地利用现有资源并提高商业可预测性。新法规引入的欧盟层面的联合临床评估的结果将作为成员国对药物价值评估和价格谈判的基础,这一举动将为制药企业以及欧盟成员国带来效率的提高。来自欧盟 12 个国家的 13 个卫生技术评估机构共同组成的欧洲卫生技术评估网络(European Network for Health Technology Assessment, EUnetHTA)进行联合临床评估。未来三年将进行评审方针和指导性文件的编撰工作,从 2025 年 1 月起将对所有肿瘤药品和前沿治疗药物(ATMPs)进行评估,2028 年起罕见病药物将被纳入评估,2030 年起所有其余领域的药物都将被纳入评估范围。 创新药物准入欧洲市场的复杂性药品作为一种特殊的商品在商业化过程中需要面临纷繁复杂的挑战,尤其在进入世界第二大医药市场——欧洲市场时更考验制药企业的战略能力。欧洲有大大小小约 50 个国家,其中 27 个国家属于欧盟成员国,30 个国家属于欧洲经济区。每个国家都有自己的国情和政策也都有不同的经济发展和购买力水平。这就使得进入欧洲市场困难重重。来源: European Federation of Pharmaceutical Industries and Associations 「千呼万唤始出来」的 欧盟统一卫生技术评估为了统一欧盟市场、降低行政成本及让创新药物更快地惠及民众,欧洲药品管理局公布了中央审批程序,通过审批的药品有权在所有欧盟国家和欧洲经济区进行上市销售。但是,后续的卫生技术评估仍需要在各个国家分别进行。虽然这考虑到了不同国家和地区的具体情况,但它也导致了商业规划方面的安全性降低以及制药公司和卫生技术评估机构的不必要的重复工作。因此,欧盟委员会和部长理事会于 2004 年呼吁欧盟统一的卫生技术评估为「潜在的优先事项」,并指出「欧洲迫切需要建立一个可持续的且统一的卫生技术评估网络」。在欧盟委员会的呼吁下,欧盟 (以及欧洲经济区) 卫生技术评估网络 (EUnetHTA) 于次年成立。欧盟委员会于 2018 年公布了一项关于在卫生技术评估框架内对卫生技术进行联合临床评估的提案。虽然一些欧盟成员国对该提案表示欢迎,但反对的声音依旧存在,特别是来自卫生评估标准较高的国家,例如,德国。经过长达三年的谈判直到 2021 年年底各方经过妥协最终达成一致意见:卫生技术评估的临床价值评估部分将在欧洲层面集中进行,非临床评估部分,例如定价和附加值程度的决定将继续在国家层面进行。可以说,该法规最重要的影响是引入了欧盟层面的联合临床评估(Joint Clinical Assessments)。这些联合评估本质上是相对有效性评估,将由指定的各个国家卫生技术专家在欧盟层面进行。但是,成员国之间达成的最终文本中表明,联合临床评估报告不具有完全的约束力,也就是说各个国家的卫生技术机构可以对总体临床效益做出自己的价值判断和结论。这意味着在联合临床评估之后,成员国仍然可以要求开发商进行澄清,并要求提供额外的证据,以进行补充的临床分析。虽然联合临床评估报告不完全具有约束力,但这并不意味着欧盟卫生技术评估条例将会成为摆设,因为成员国卫生技术机构通过协调小组都有直接参与欧洲层面的临床评估。因此,在实践中,就成员国层面的报销程序而言,要想在国家层面对欧盟层面的积极的联合评估结论提出质疑,需要相当大的说服力。 EUnetHTA 的组成以及时间规划EUnetHTA 21 联合体由 ZIN(荷兰)领导的以下国家卫生技术评估机构组成:AEMPS (西班牙),AIFA (意大利),AIHTA (奥地利),G-BA (德国),HAS (法国),INFARMED (葡萄牙),IQWIG (德国),KCE (比利时),NCPE (爱尔兰),NIPN (匈牙利),NOMA (挪威),TLV (瑞典),ZIN (荷兰) 。来源:EUnetHTA2022 年 1 月 11 日,欧盟出台了新法规《欧洲卫生技术评估》:2022-2025 为期三年的时间用来制定和部署具体的科学评审规则。自 2025 年起,所有肿瘤药品和前沿治疗药物(ATMPs)必须经过联合临床评估,2028 年起,所有罕见病药物也将纳入评审中,2030 年起评审将包含所有其他的药品。来源:笔者自己做的考虑到这一时间表,预计在 2025 年获得欧洲批准的医药产品的临床 II 期和/或 III 期研究,应该按照欧盟卫生技术评估条例的新要求进行规划。鉴于许多指导性文件仍未公布,参加联合科学咨询会议(Joint Scientific Consultation)就显得尤为重要。联合科学咨询将为制药企业在关键性临床试验开始之前(在可行性/概念证明研究之后)提供不具约束力的科学建议,以提高数据的质量和适当性,以便于未来的卫生技术评估。此外,通过联合科学咨询会议可以让制药企业和卫生技术评估机构及监管机构(欧洲药品管理局)在药品研发的早期阶段进行交流,以便在研究设计(关键性试验和上市后证据生成)和经济证据生成计划中整合多个欧洲成员国的不同要求(如选择比较者、相关结果、生活质量、患者群体)。在没有具体评审方针和相关法规的情况下,EUnetHTA 21 联盟被指定在 2023 年 9 月前进行 6 至 8 个医药产品的联合科学咨询会议。第一次公开征集已有三个医药产品被选中参加咨询会议。第二次公开征集也已经于 2022 年 8 月 31 日结束,此次征集将在 9 月选出剩余最多 5 个医药产品进行咨询会议。所有成果、公众咨询时间表和草案的最终定稿都将在 EUnetHTA 网站上定期更新。 欧盟统一卫生技术评估的展望欧盟卫生技术评估条例被认为是欧盟制药战略的第一个成果,其公开的目标是促进患者获得创新和可负担得起的药物,并支持欧盟制药业的竞争力和创新能力。该法规对我国创新药企进入欧盟市场来说既有利好,同时也充满了挑战。利好之处在于,首先是语言,EUnetHTA 用英文作为官方使用语言,所有的沟通和需要提交的药物评估档案均使用英语,相较于之前在每个国家都要准备一份当地语言的药物档案用于评审来说极大的节约了人力成本和沟通成本。此外,从评审方法上来说,为了照顾到各个成员国的情况,评估的方法和要求可能会较为宽泛,取得有利的评审结果可能性可能会更大。挑战之处在于很大的不确定性和未知性。例如联合评审小组如何执行新的评估方法和要求,在遇到特例或复杂情况时该如何处理,面对成员国意见无法达成一致时该如何解决,一切的一切都是在摸着石头过河。但总体来看,机遇都是大于挑战的。未来,如有需要,笔者也会分析并更新 EUnetHTA 出台的重要的公告和评审相关的规则。参考文章:https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32021R2282https://www.eunethta.eu/点击卡片进入 Insight 小程序药品申报、临床、上市、一致性评价…随时随地查!👇👇👇免责声明:本文仅作消息分享,并不构成投资建议,也不代表 Insight 数据库的立场,文章观点仅供分享行业见解,请广大投资者谨慎。编辑:HebePR 稿对接:微信 insightxb投稿:微信 insightxb;邮箱 insight@dxy.cn点击阅读原文免费试用 Insight 数据库
医药出海
2022-10-27
本次直播免费参加,点击上方链接马上报名!过去两个月,蓝鸟生物接连在美国推出两款基因疗法,定价更是一路走高,最贵达到300万美元。受此影响,蓝鸟生物萎靡不振的股价终于有了一点起色。但值得注意的,无论是Skysona还是Zynteglo,此前皆已在欧洲市场获批,商业化却并不理想。去年,蓝鸟生物由于跟支付方的谈判失败等因素,决定撤出欧洲市场,转道美国。蓝鸟生物的困境并不是个案,数据显示,不少基因疗法的制造商也相继在欧盟受挫。在这篇文章中,Research Partnership回顾了为什么过去几年欧洲六项批准的基因疗法商业化之路并不顺利,并分享从其支付网络收集到的关于欧洲基因治疗市场准入前景的反馈。12022年成为创纪录之年?今年1月,再生医学联盟(ARM)的《细胞和基因行业状况》简报,将2022年指定为罕见疾病基因疗法批准的创纪录年份。但这并没有发生。截至今年9月,ARM名单上仅有三种基因疗法获得了EMA的批准,其中三种有望在2023年进入市场。基因疗法的开发、制造和给病人使用都是复杂而昂贵的。基因疗法与高昂的材料成本相关,因此需要一个能反映其治疗价值的价格。对于超罕见病,受益的患者数量少,意味着需要高昂的价格来确保盈利,而事实证明,实现这一点具有挑战性。虽然该行业仍然积极开发包括血液病和眼疾在内的罕见疾病领域的先进疗法(包括基因疗法),但从整个先进疗法部门向更普遍的疾病转移的趋势越来越明显。根据ARM的数据,在2405项先进治疗临床试验(包括行业和学术/政府资助的研究)中,只有59%的研究对象是ARM定义为“常见”的疾病。2基因疗法在路上“颠簸”关于基因疗法的最新积极消息中,夹杂着一系列“又来了”的时刻。2022年7月,意大利非营利组织Telethon基金会关于腺苷脱氨酶严重联合免疫缺陷症(ADA-SCID)的Strimvelis的决定,再次突显了一种超罕见病基因疗法商业化带来的经济挑战。当前许可证持有者Orchard Therapeutics由于缺乏经济可行性而选择退出该项目后,基金会决定独自负责Strimvelis的商业化。下图显示了细胞和基因治疗在欧洲市场开拓的曲折历史。在Strimvelis开发之前,美国蓝鸟生物高调决定停止在欧洲的业务,原因是该公司与报销当局就其β-地中海贫血基因疗法Zynteglo进行了两年的谈判失败。Zynteglo未能获得适当的价值认可,且无法确定商业化合作伙伴,导致该公司还撤回了其在欧洲的脑肾上腺脑白质营养不良(ALD)基因疗法Skysona的市场监管许可。Skysona是2021年EMA批准的唯一一种基因疗法。当Telethon基金会介入拯救Strimvelis时,它回应了许多开发者的想法,称考虑到基因疗法与传统药物相比的独特性,科学进步还没有与基因疗法的监管、市场准入过程的充分发展相匹配。基因疗法面临着一系列的证据生成挑战,包括治疗效果的大小和持续时间的不确定性、定义不清的患者群体、试验持续时间和规模的限制以及对单臂试验和替代终点的依赖。与表征价值相关的挑战,使Research Partnership对卫生技术评估(HTA)机构和支付方应对即将涌入的具有固有高标价的基因疗法的准备情况感到好奇。从Research Partnership的全球网络中,研究人员联系了来自英国、法国、德国、西班牙和意大利的七个拥有基因治疗专业知识的支付方,以征求他们的看法。当被问及他们国家的定价和补偿(P&R)系统的准备情况时,大多数支付方都认为,他们在一定程度上做好了准备。这种准备程度,部分与其对收集真实世界证据(RWE)的重要性的认识有关,这可以在启动时解决证据的不确定性。受访者普遍觉得,RWE有一定的重要性。然而,一个法国支付方强调,这取决于目标和背景。法国卫生咨询机构Haute Autorité de santé’s (HAS’s )对基因疗法的最新评估,要求RWE收集作为重新评估的一部分,包括监测数据作为早期访问计划的一部分。德国支付方指出,德国联邦联合委员会(G-BA)会选择某些EMA有条件批准的产品和孤儿药,即必须作为强制RWE数据收集注册的一部分的产品。虽然预计只针对选定的产品,但注册要求旨在帮助克服上市时证据不足带来的不确定性。在第一个测试案例后,治疗脊髓性肌肉萎缩的Zolgensma和A型血友病的Roctavian是下一个接受注册的基因疗法。3Zynteglo退出欧洲的影响大多数接受采访的支付方预计,Zynteglo的挑战将对未来其他基因疗法的P&R评估产生中度至高度的负面影响。只有三个受访者(其中两个来自法国)认为此举不会产生影响。一个法国支付方强调,对于12岁以上至35岁以下的患者,HAS授予Zynteglo III级临床疗效改善状况(ASMR)评级。这意味着它处于有利地位,可以从法国卫生产品经济委员会(CEPS)获得高价,直到谈判因上市许可撤回而停止。德国支付方认为,Zynteglo的标价是157.5万欧元,这是导致该公司商业化失败的主要原因之一。在德国《医药产品市场的改革法案(AMNOG)》仲裁委员会的审理过程中,考虑到每年输血的成本非常低,这个价格是不合理的。在蓝鸟生物无法与德国国家法定健康保险基金协会(GKV-SV)就最终补偿价格达成一致后,仲裁委员会召开了会议。根据欧洲制药企业家联合会(EUCOPE)的说法,这不是G-BA的错,而是GKV-SV在不承认基于绩效的担保方面采取的强硬路线。4年金支付在某些地区落地关于创新的付费模式以更好地反映基因治疗的价值,已经有了相当多的讨论,但讨论的数量没有与实施相匹配。找到一种方法来真正实现它们的价值并克服不确定性,这一点仍然具有挑战性。随着更多基因疗法的涌入,与下表中列出的其他模式相比,Research Partnership调查的支付方对以下模式更加开放,包括基于结果的支付(非年金)、年金支付(基于结果)和价格数量协议。基于结果的支付(非年金)模型,在德国已被个人疾病基金用于迄今商业化了的基因疗法。GKV-SV层面上,对通过强制注册表数据收集进行证据开发的产品,更常见的是使用一次性支付。在德国和法国,年金支付(基于结果)模式正在成为可能,这种模式将允许基因治疗的高额前期费用在多年中分摊。截至2022年3月,德国国内围绕修改GKV-FKG法案2021年高成本治疗风险池机制(取消分期付款合同的激励机制)展开了争论。而法国方面,2021年3月出台的CEPS-创新药品行业协会LEEM框架协议,包括了合同修正案,可能会在几年内实现分摊支付,但这些修正案尚未落实到立法中。5开发跨境通道的重要性除了国家市场准入挑战外,特别是在超罕见领域,制造商还必须通过欧盟的跨境报销途径。根据该途径,欧盟公民有权在任何欧盟国家获得医疗保健服务,并由其母国报销医疗费用。由于患者管理的复杂性和所需的临床专业知识,这一途径正在成为治疗严重遗传和超罕见疾病的产品的关键市场准入途径,对于这些疾病,往往需要优质的专家中心。尽管英国脱欧,但在2022年2月,Orchard宣布,患者将能够利用跨境途径从英国的中心(欧洲五个中心之一)获得其基因疗法Libmeldy。尽管像Orchard这样的药企在使用这个框架,但业界认为它并不适合。关键障碍是复杂性和不透明的自由裁量批准过程,特别是对于在患者所在国没有良好P&R结果的产品。行业似乎还需要更好的规划。在Research Partnership的研究中,七个支付方中只有两个(分别来自德国、法国)表示,他们的国家为管理这一途径做好了充分准备。有趣的是,欧盟新的联合临床评估计划于2025年开始对先进疗法进行评估,但接受采访的支付方并不指望这能够帮助跨境途径充分发挥其潜力。一个法国支付方称,这是因为P&R决策将继续在全国范围内进行。6需要政策回应的思维转变在跨境考虑方面,制药产业有必要继续进行游说,要求提高透明度和理解,以便简化罕见病的基因疗法的指导,作为另一种市场准入途径。尽管为2025年欧盟HTA先进疗法的节点做准备是至关重要的,但国家P&R决定将继续左右着基因疗法的商业命运。Strimvelis的案例也强调,需要一个政策环境来支持在罕见疾病的基因治疗方面取得的进展。这离不开有关该领域的经济挑战的认识,此类挑战对定价有影响。重要的是,基因治疗的前景可以在罕见病适应症中得到满足,尽管最终价值可能更多地取决于常见疾病的适应症。在追求这些适应症的过程中,基因治疗商业化代表着一场“极端风暴”,因为经济可持续性的问题变得尖锐。Research Partnership认为,除了潜在的高预算影响之外,β-地中海贫血和血友病等疾病的未满足需求,并不比“天价药”Zolgensma治疗的脊髓性肌肉萎缩症更高。此外,在这种情况下,对照药物的价格通常要低得多。这意味着,HTA机构和支付者将继续要求更多的数据,来证明报销这类基因疗法的合理性。因此,正如英国的一项调查所强调的那样,对于药企而言,从这些利益相关者那里寻求数据要求和期望的早期反馈是很重要的。这些反馈应该涉及一系列方面,包括临床试验设计、经济建模计划、额外的数据收集要求和合适的支付模式。为了提高启动时的HTA/P&R结果,关键是要加强证据生成计划,以显示潜在的药效终生持久性和获取额外的价值元素。承诺长期持续提交上市后数据、后续关键临床试验数据和RWE收集,可以支持这方面的工作。临床试验中与健康相关的生活质量的直接测量,也有助于证明生活质量的改善。持续进行HTA机构和支付方教育,对改变思维方式是必要的,特别是在涉及常见疾病的治疗时。这将有助于增加支付意愿和支持,推动更大的HTA/P&R流程调整,同时减少对预算影响、卫生系统可持续性和行政负担的担忧。创新的支付模式,例如基于结果和年金的模式,是确保收集相关数据的潜在解决方案,以减少支付方对这些疗法有效性的长期不确定性,同时提供合理的投资回报。然而,这种模型的开发和实现是复杂的。支付方还可以通过将其费用分摊到与成果展示相关的几年,进而解决可负担性/预算影响问题,但这有赖于更大的灵活性和立法改革。在获得EMA有条件批准后,所有人都在关注Roctavian如何在欧盟P&R领域取得成功。参考文献:1.定价300万美元!全球最贵基因疗法会让蓝鸟生物再次腾飞吗?2.风口起飞:基因疗法的价格会越来越高吗?3.写意讨论|基因治疗药物开发的未来之路41.Gene therapies for prevalent diseases in Europe – the perfect storm of economic sustainability?;pharmaphorum
基因疗法创新药合作孤儿药细胞疗法
100 项与 Hassab Labs 相关的药物交易
登录后查看更多信息
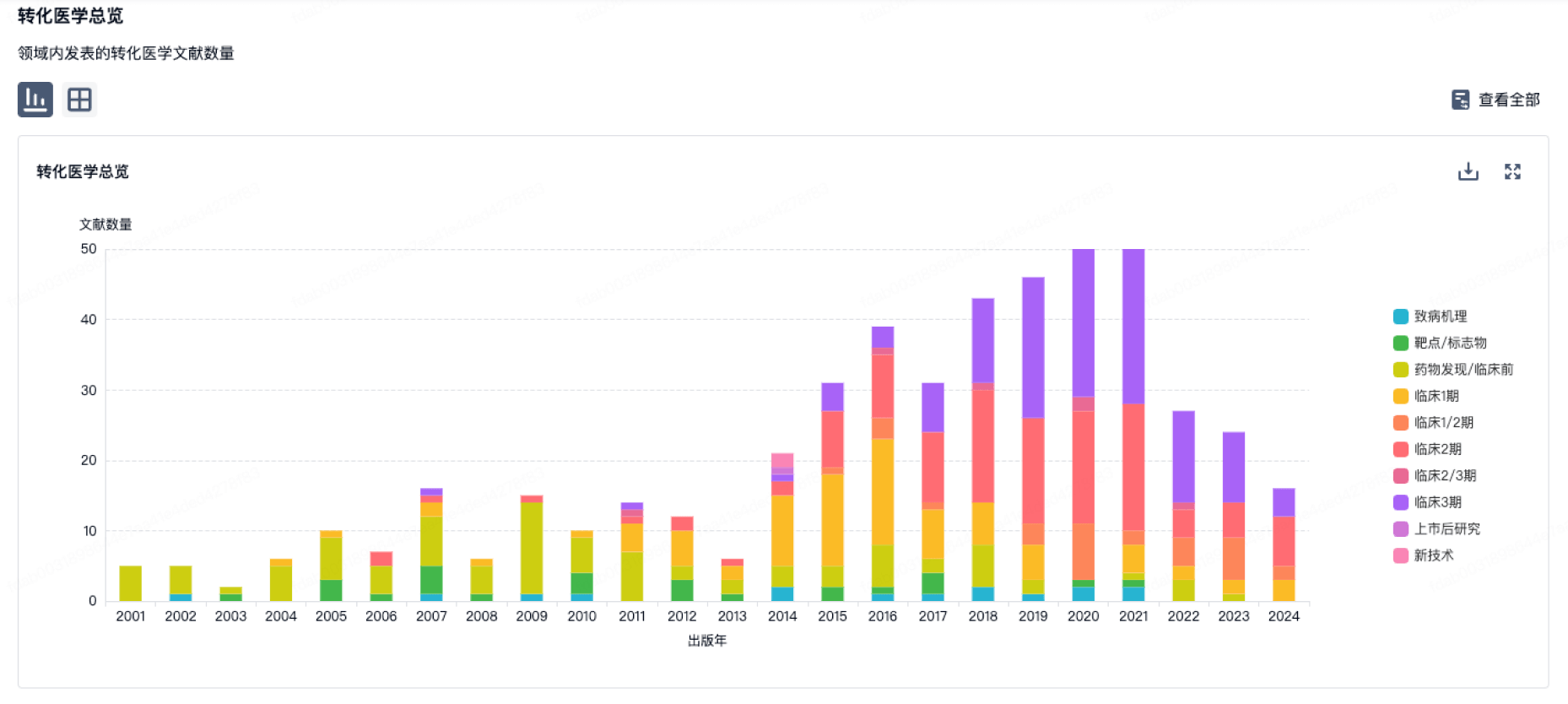
100 项与 Hassab Labs 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年07月08日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
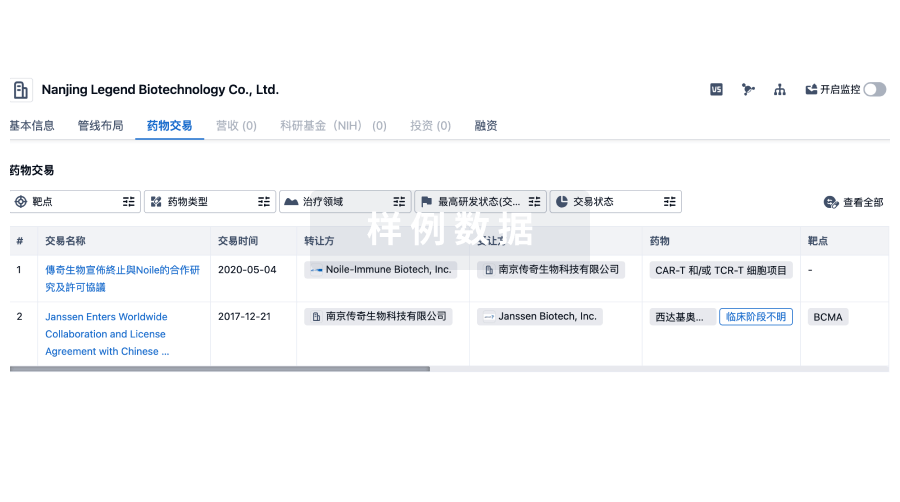
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
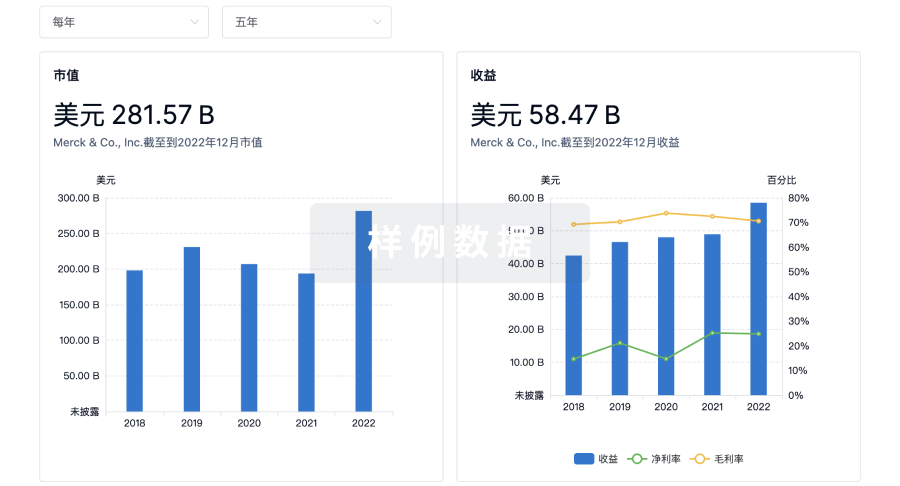
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
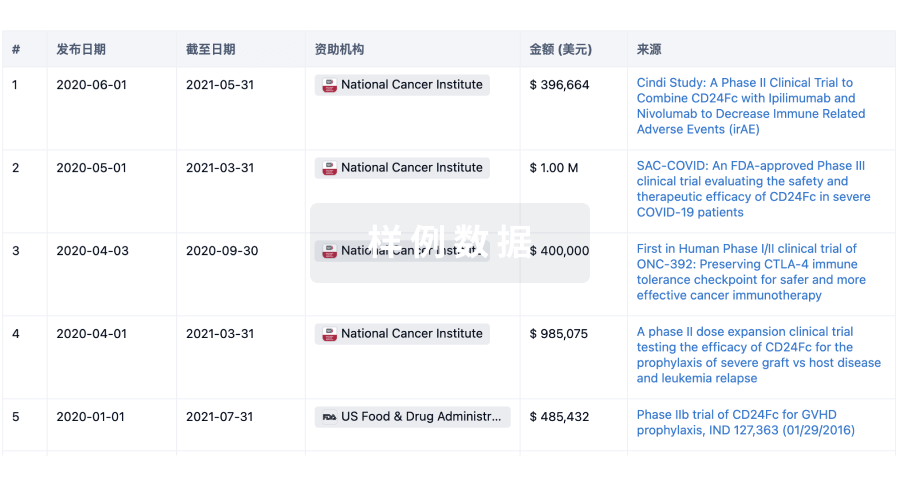
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用