预约演示
更新于:2025-03-20
Potrasertib
更新于:2025-03-20
概要
基本信息
原研机构 |
在研机构 |
非在研机构- |
最高研发阶段临床1期 |
首次获批日期- |
最高研发阶段(中国)临床1期 |
特殊审评- |
登录后查看时间轴
结构/序列
分子式C28H30Cl2N8O |
InChIKeyBNVDJVMTLDUCGH-HDICACEKSA-N |
CAS号2226938-19-6 |
关联
2
项与 Potrasertib 相关的临床试验CTR20212068
一项评估WEE1抑制剂IMP7068片单药治疗晚期实体瘤患者的安全性、耐受性、药代动力学和抗肿瘤活性的I期、开放、多中心、剂量递增和扩展研究
剂量递增阶段主要目的:
-评价晚期实体瘤患者单次和重复给予IMP7068片的安全性和耐受性;
-确定IMP7068片单药治疗的最大耐受剂量(MTD)和II期推荐剂量(RP2D);
次要目的:
-表征IMP7068片在晚期实体瘤患者中单次和重复给药的药代动力学(PK);
-初步评估IMP7068片重复给药在晚期实体瘤患者中的抗肿瘤活性; 剂量扩展阶段主要目的:
-评价IMP7068片单药治疗的抗肿瘤活性;
次要目的:
-使用其他疗效指标评价IMP7068片单药治疗的抗肿瘤活性;
-评价IMP7068片单药治疗的安全性和耐受性。
开始日期2021-12-27 |
申办/合作机构 |
NCT04768868
A Phase 1, Open-Label, Multi-Center, Dose Escalation and Expansion Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Anti-Tumor Activity of the WEE1 Inhibitor IMP7068 Monotherapy in Patients With Advanced Solid Tumors
A Phase 1 Dose Escalation and Expansion Study of IMP7068 Monotherapy in Advanced Solid Tumors
开始日期2021-02-25 |
申办/合作机构 [+1] |
100 项与 Potrasertib 相关的临床结果
登录后查看更多信息
100 项与 Potrasertib 相关的转化医学
登录后查看更多信息
100 项与 Potrasertib 相关的专利(医药)
登录后查看更多信息
14
项与 Potrasertib 相关的新闻(医药)2025-01-30
近年来,互联网“一哥”腾讯在制药行业的投资事件可以说是越来越多,从最早期更偏向于互联网的药品电商平台,或者是诊断平台,到中期开始关注生产制造商,最近几年来也兴起了投资Biotech的热情,本文将简单汇总近几年来腾讯在Biotech企业中的具体投资项目。
Atomwise
Atomwise是一家成立于2012年的AI制药公司,腾讯参与了其A轮和B轮融资,不过目前该公司管线还在早期开发过程中,并没有太多进展信息。
礼邦医药
2018年成立的礼邦医药(Alebund)专注于针对肾脏疾病及其并发症以及其他慢性疾病的新疗法的发现、开发、生产和商业化。目前礼邦医药已经针对一系列肾脏适应症开发了相应管线,包括慢性肾脏疾病(CKD)/透析并发症、IgA肾病、糖尿病性肾脏疾病、局灶节段性肾小球硬化(FSGS)和常染色体显性多囊肾病(ADPKD)。
公司在研管线中进展最快的是AP301。这是一种同类最佳的铁基磷结合剂,主要用于治疗慢性肾脏病患者的高磷血症。AP301具有优异的药代动力学特性和安全性,能够显著降低患者的血磷水平并改善生活质量。目前,AP301正在中国进行新药上市申报,并计划在全球开展注册三期临床试验。
另外公司对外引进了罗氏的美信罗,美信罗是全球首个获批的持续性促红细胞生成素受体激动剂,礼邦医药将负责美信罗在中国大陆地区的商业化。
腾讯投资与熙诚金睿等机构共同参与了礼邦医药的C轮融资,该轮融资额达到了5.5亿元人民币。
圣因生物
圣因生物是一家致力于开发基于RNA干扰(RNAi)技术的新型小核酸药物的生物制药公司,在中美均建有研发中心。
其中,A+轮融资超八千万美元(近6亿元人民币),由腾讯投资和元生创投领投,北极光创投、建发新兴投资、元禾控股、上海生物医药基金、深创投、泰达科投等机构跟投。老股东启明创投、雅惠投资、险峰旗云、泰福资本、临港蓝湾资本也持续加持。
目前公司管线主要处于临床早期,临床进展最快的管线是靶向PCSK9的siRNA-GalNAc 结合物SGB-3403。
宁丹新药
宁丹新药是一家聚焦于中枢神经系统疾病(CNS)领域新药研发和产业化的创新型公司,是先声系的药企,同时还兼具CDMO服务业务。
旗下研发平台可以提供药物化学合成、化合物筛选、CMC开发、DMPK研究、体内药理研究、毒理研究和临床研究等综合服务。管线状况如下图所示:
目前,该公司已有与先声药业和北京天坛医院合作的创新药先必新(研发代码:Y-2)在我国上市,适应症为急性缺血性脑卒中,是全球首个中风急救药,在美国也被授予了突破性疗法认定。
先必新由依达拉奉和右莰醇两种活性成分以4:1的配比组合而成。依达拉奉通过清除自由基,保护神经细胞免受氧化应激损伤。右莰醇则具有抗炎作用,能够抑制炎症反应的发生和发展。一方面,依达拉奉和右莰醇分别通过清除自由基和抑制炎症反应来保护神经细胞免受损伤;
另一方面,这两种成分还能够相互协同,增强彼此的保护作用,从而更全面地保护神经细胞并促进其功能恢复。
跃赛生物
跃赛生物是一家专注于开发新一代基于人多能干细胞的细胞治疗药物的创新型制药公司。该公司自2021年创立以来,已经开发了多个关键技术平台,包括重编程技术平台、干细胞分化平台、SISBAR谱系示踪技术以及高精度基因编辑平台等。
跃赛生物的研发管线布局以神经退行性疾病为核心,今年12月27日,治疗帕金森病的自体细胞注射液UX-DA001已经获得了国家药品监督管理局药品审评中心(CDE)的临床试验默示许可。这是中国第一款、全球第二款进入临床阶段的针对帕金森病的自体iPS衍生细胞治疗药物。
英派药业
英派药业是以开发DNA损伤修复通路(DDR)为核心的制药公司,公司产品管线包括PARP抑制剂(senaparib/IMP4297)、Wee1抑制剂(IMP7068)以及多个其他DDR靶点抑制剂。
其中senaparib已获得中国国家药品监督管理局(NMPA)批准上市,用于晚期上皮性卵巢癌、输卵管癌或原发性腹膜癌成人患者在一线含铂化疗达到完全缓解或部分缓解后的维持治疗。
2024年11月,根据天眼查显示英派药业发生工商变更,新增广西腾讯创业投资有限公司为股东。
华毅乐健
华毅乐健创立于2019年,由著名生物学家饶毅教授创立。目前该公司在肝脏相关疾病,神经中枢系统疾病等疾病领域上建立了8条产品管线,构建了罕见病与常见病相平衡的产品管线布局。
华毅乐健的先导产品是针对血友病A的AAV疗法GS1191,是国内首个进入探索性临床试验阶段的产品,也是国内首个获批IND的血友病A基因治疗产品。
该产品通过肝脏基因递送技术,将人凝血因子FVIII基因导入患者体内,实现长期、稳定的凝血因子VIII表达,从而治疗血友病A。
圆音生物
圆因生物科技有限公司成立于2021年4月,专注于环状RNA技术在创新药物和创新疗法领域的研究和应用。2022年4月企查查显示,广西腾讯创业投资有限公司以8.67%的股份入股了该公司。目前该公司研发管线大都处于临床早期。
极目生物
极目生物是一家总部位于中国、专注于眼科疗法的生物技术,拥有从早期发现阶段到商业化阶段的一系列突破性眼科治疗技术产品组合。
值得一提的是,该公司除了眼科治疗药物外,还有眼科治疗器械iTEAR睦沁上市,这是一款通过鼻外神经刺激促进自身天然泪液分泌的二类医疗器械。它采用物理振动的方式在鼻外部进行刺激,激活鼻泪反射,最终作用于泪腺、睑板腺和杯状细胞,分别促进水液、脂质和黏蛋白的分泌,即促进包含这三大重要成分的天然泪液的分泌。
除了iTEAR睦沁之外,极目生物不仅拥有脉络膜上腔注射创新给药技术,角膜内皮细胞疗法、青光眼神经保护基因疗法等前沿科技,也差异化布局了包括针对近视、老视的微矩阵药膜递送疗法在内的消费者导向创新技术。
2021年3月10日,眼科生物技术公司极目生物宣布完成超1亿美元B轮融资。本轮融资由正心谷资本领投,腾讯、Octagon Capital和企业家及新世界集团联席行政总裁郑志刚博士参投,原股东南丰生命科技、鼎丰生科资本、晨兴创投继续加持。
晟斯生物
晟斯生物成立于2020年,是一家主要以血友病药物开发为核心,向大分子生物制药药企转型的罕见病药物开发公司。目前该公司兼有四款长效/超长效重组凝血因子开发管线,覆盖血友病A、血友病B、抑制物阳性等血友病主要适应症,并将拓展到急性止血、创伤止血等适应症。
在D轮融资中,腾讯投资和张江科投投资了该公司数亿元。
信诺维
信诺维成立于2017年,是一家兼具科研实力和商业化能力的平台型创新药公司。基于自主创新的靶向治疗、抗感染和PROTAC三大技术平台,信诺维已形成BLI抑制剂、EZH2抑制剂、hURAT1抑制剂以及新一代ADC等创新药管线,覆盖肿瘤、抗感染和代谢等疾病领域。
在进度较快的管线中,XNW4107是信诺维自主研发的新一代β-内酰胺酶类广谱抗耐药菌药物,对于近年来逐年增加的碳青霉烯耐药的细菌感染具有强有力的抗菌疗效,已经进入Pre-NDA申请阶段。XNW3009是新一代降尿酸药物,降尿酸效果远优于对照组;XNW5004是新一代EZH2抑制剂,在血液瘤、前列腺癌等适应症中已显示出优秀的抗肿瘤疗效。
2024年2月信诺维完成了7亿元E轮融资,腾讯投资和国鑫投资联合领投。
总结
总的来看,腾讯在近几年的商业化投资,还是呈现出向临床更早期企业发展的情况,对于投资的数额也呈现出逐年增加的态势,投资企业赛道较为多样化。
参考来源:
各个公司官网
突破性疗法siRNA临床3期临床申请引进/卖出
2025-01-21
·小药说药
关注小药说药,一起成长!
前言
合成致死性是一种遗传现象,即单个基因缺陷与细胞存活相容,但两个基因的同时缺陷导致细胞死亡或细胞适应性受损。合成致死性提供了一个独特的机会,可以间接靶向以前被认为“难以成药”的蛋白质,包括携带功能丧失(LOF)突变的关键肿瘤抑制蛋白和由扩增或其他基因组改变引起的过表达致癌蛋白。这些与环境相关的相互作用是癌细胞固有和特异性的,因此提供了选择性靶向这些细胞的机会,同时保留了缺乏相关基因组背景的非癌细胞。
目前,PARP抑制剂已经在BRCA突变癌症患者中获得临床成功。在此推动下,针对DNA损伤反应途径中多种合成致死相互作用的新型药物正在临床开发中,针对跨越表观遗传、代谢和增殖途径改变的合成致死相互作用的合理策略也已出现,并进入临床前和早期临床试验阶段。
靶向合成致死相互作用
DNA损伤反应(DDR)网络可以保持基因组稳定性,抑制复制应激,保护受损的复制叉,并确保高保真DNA复制。DDR成分的遗传性和获得性细胞缺陷与基因组不稳定性的积累有关,导致肿瘤发生和癌症进展。同时,具有这些缺陷的细胞变得依赖于补偿DDR途径生存,从而为合成致命靶向创造了机会。
DNA损伤信号:靶向ATR、ATM和DNA-PK
ATR、ATM和CHK1激酶的抑制剂主要通过抑制必需的细胞周期检查点来利用细胞复制应激,导致过早进入有丝分裂和细胞死亡。ATR是一种丝氨酸/苏氨酸蛋白激酶,属于磷脂酰肌醇3-激酶相关激酶(PIKK)家族。当DNA发生损伤时,ATR被激活,并通过下游信号通路参与DNA损伤的检测、复制叉的稳定和损伤修复;激酶ATM通过调节DDR内下游激酶的活性和控制细胞周期检查点进程,在DNA双链断裂(DSB)的修复中起着关键作用;DNA-PK是另一种磷脂酰肌醇3-激酶相关激酶(PIKK),在DNA损伤部位充当邻近修复蛋白的蛋白质支架,在非同源末端连接(NHEJ)中起着关键作用。许多肿瘤细胞由于DNA修复通路的缺陷,高度依赖这些激酶途径来维持其生存和增殖。因此,ATR、ATM和DNA-PK抑制剂与这些缺陷相结合,可导致肿瘤细胞发生合成致死。
多种ATR抑制剂已处于临床开发中,包括berzosertib、ceralasterib、elimusertib、camonsertib、tuvusertib、ART0380、ATRN-119、ATG-018和IMP9064等;正在临床试验中测试的ATM抑制剂包括AZD1390和双重ATM和DNA-PK抑制剂XRD-0394和M4076,临床开发中的DNA-PK抑制剂包括AZD7648和peposertib。
DNA复制和细胞分裂:WEE1、PKMYT1、CDC7和PLK4
Wee样激酶1(WEE1)通过催化CDK1和CDK2在Tyr15位置的抑制性磷酸化来调节细胞周期通过G2/M和S检查点的进程。WEE1的抑制在G1/S失调的情况下是合成致死的,例如由CCNE1扩增或TP53突变引起的G1/S失调。正在临床试验中测试的WEE1抑制剂包括adavosertib、azenosertib、Debio 0123、IMP7068、SY-4835、SC0191和ATRN-W1051。
蛋白激酶,膜相关酪氨酸-苏氨酸1(PKMYT1)是一种细胞膜相关丝氨酸-苏氨酸蛋白激酶,也调节CDK1和G2/M检查点。功能基因组筛选的数据表明,PKMYT1失活与CCNE1扩增具有合成致死性。此外,除了CCNE1扩增,编码E3泛素连接酶FBXW7基因中的LOF突变和编码PP2A磷酸酶亚基PPP2R1A的基因也可通过PKMYT1抑制靶向。
细胞分裂周期7(CDC7)激酶在触发复制起点激活中起着重要作用。携带功能获得TP53突变的细胞依赖于CDC7依赖性DNA复制,这是由于与致癌转录因子MYB合作促进癌症细胞中的CDC7激活。在携带TP53突变的癌症细胞中,CDC7的药理学抑制诱导选择性衰老,而mTOR信号与CDC7联合抑制在促进凋亡细胞死亡方面非常有效。然而,尽管具有明显的临床前活性,但几种CDC7抑制剂(BMS-863233、TAK-931、NMS-1116354和LY3143921)在早期试验中未能显示出足够的疗效。
Polo 样激酶4(PLK4)是一种调节中心粒生物发生的激酶,中心粒是有丝分裂纺锤体组装和染色体分离所需的中心体的重要组成部分。中心粒周围的蛋白质是另一个关键的中心体成分,受E3泛素连接酶TRIM37的负调控。TRIM37的扩增(通常在神经母细胞瘤和乳腺癌中观察到)导致中心体物质耗竭,从而增加了对中心粒进行有丝分裂纺锤体组装的依赖,并使PLK4抑制在这种情况下成为一种有吸引力的治疗策略。目前,两种PLK4抑制剂CFI-400945和RP-1664正在临床试验中进行测试。
DNA修复:WRN、POLQ和USP1
POLQ和泛素特异性蛋白酶1(USP1)是携带BRCA1/2缺陷的癌症中临床相关的合成致死靶点。POLQ是一种广泛保守的DNA聚合酶,也是DSB修复过程中易出错的MMEJ途径的核心介体。来自几项试验的数据表明,POLQ抑制和BRCA1/2缺乏之间存在合成致死关系,这归因于癌症HRR缺陷(HRD)细胞对MMEJ
DNA修复的高度依赖性。测试各种POLQ抑制剂的早期试验正在进行中,包括ART4215、novobiocin、ART6043和GSK4524101,作为单一疗法或与各种PARP抑制剂联合使用。
USP1是一种去泛素酶,调节关键的范可尼贫血复合物和跨损伤合成底物,同时抑制NHEJ。在BRCA1缺陷细胞中,USP1在复制叉处特别活跃,其与叉DNA结合并被其激活,同时介导叉保护;抑制USP1会导致复制叉失稳、叉保护失败和细胞死亡。KSQ-4279是一种USP1抑制剂,目前正在I期试验中作为单一疗法或与PARP抑制剂或铂类化疗联合使用进行测试。
Werner综合征ATP-依赖性解旋酶(WRN)功能的丧失与癌细胞中的微卫星不稳定性-高(MSI-H)表型具有合成致死关系。WRN敲除已被证明会在MSI-H细胞中诱导广泛的DSBs、细胞周期失调、基因组不稳定和凋亡,但在体外微卫星稳定细胞中不会诱导。WRN抑制剂HRO761和RO7589831均已进入I期临床试验。
靶向合成致死代谢依赖性
代谢重编程是癌症的标志,肿瘤代谢的遗传或表观遗传改变,是调节细胞内自由基积累和维持肿瘤进展所需的生物合成和能量需求增加所必需的。甲硫基腺苷磷酸化酶(MTAP)缺失与PRMT5-MAT2A-RIOK1轴成分耗竭之间具有合成致死关系。MTAP的损失导致5′-甲硫腺苷(MTA)的积累,MTA本身通过竞争PRMT5
S-腺苷甲硫氨酸(SAM)结合袋,成为PRMT5的强效和选择性内源性抑制剂。因此,缺乏MTAP功能的细胞PRMT5甲基化水平降低,对进一步的PRMT5耗竭敏感,导致细胞生长抑制。目前正在临床试验中测试几种MTA协同PRMT5抑制剂,如AMG193、MRTX1719和TNG908,以及MAT2A抑制剂IDE397。
涉及代谢途径的合成致死相互作用还包括氨基酸和核酸代谢途径中的合成剂量致死性关系。例如,FLT3内部串联重复(FLT3-ITDs)是驱动因素,通过激活参与从头丝氨酸生物合成基因的转录因子4(ATF4)依赖性转录调控,赋予丝氨酸生物合成独特的代谢依赖性。在这种情况下,抑制丝氨酸生物合成中第一个也是唯一的限速酶磷酸甘油酸脱氢酶(PHGDH),使用PHGDH抑制剂WQ-2101在FLT3野生型细胞中亚致死的剂量,会导致凋亡诱导。与FLT3野生型细胞系相比,对携带FLT3-ITD突变的细胞系具有显著的抗增殖作用。
靶向表观遗传调控的合成致死策略
染色质调节过程的动态控制对细胞功能至关重要。在多达50%的癌症中检测到与读取、添加或删除表观遗传修饰以及影响染色质调节和组织有关的基因突变。SWI/SNF染色质重塑复合物(CRC)是四个ATP依赖性CRC家族之一,能够通过驱逐、动员或沉积核小体来改变染色质结构。编码SWI/SNF成分的基因突变大多导致功能丧失,在所有人类癌症中发生率高达20%。功能基因组筛查已证明SWI/SNF同源对SMARCA4-SMARCA2、ARID1A-ARID1B、SMARCA4-AMARCB1、SMARCA4-ARID2、SMARCA-4-ACTB和SMARCC1-SMARCC2之间存在合成致死相互作用。
作为表观遗传合成剂量致死性的另一个例子,研究发现SWI/SNF-BRG1-SMARCA4复合物在弥漫性中线胶质瘤患者中与H3K27M突变的表观遗传依赖性。SMARCA4与SOX10在H3K27M突变胶质瘤细胞的调节元件上共定位,以调节参与细胞增殖和细胞外基质的基因表达。H3K27M的功能缺失导致SMARCA4染色质在SOX10和H3K27乙酰化标记的增强子上的结合减少。使用靶向蛋白降解剂或小分子抑制剂靶向SMARCA4,在体外和体内模型中产生了抗肿瘤活性。
小结
合成致死作为一种新兴的肿瘤治疗策略,近年来备受关注。它基于两个非致死性基因同时受到抑制或干扰时,会导致细胞死亡的现象。合成致死的发现为癌症治疗提供了新的思路,推动了相关药物的开发。在合成致死策略中,PARP抑制剂是最成功的案例之一,PARP抑制剂通过抑制PARP的活性,导致DNA损伤无法修复,从而引起肿瘤细胞死亡。目前,PARP抑制剂已被批准用于治疗携带BRCA1或BRCA2基因突变的乳腺癌和卵巢癌。
除了PARP抑制剂,研究人员还在积极探索其他合成致死靶点,如ATR、PRMT5等。这些靶点的抑制剂在克服肿瘤耐药性、提高治疗效果方面显示出良好的潜力。与此同时,人们也正试图通过联合用药策略,如将PARP抑制剂与其他抗肿瘤药物联用,以提高治疗效果。总之,合成致死作为一种具有前景的肿瘤治疗策略,为癌症患者带来了新的希望。随着科研技术的不断发展,相信未来会有更多基于合成致死原理的新型抗肿瘤药物问世,为癌症治疗带来革命性的变革。
参考文献:
1.Synthetic lethal strategies for the
development of cancer therapeutics. Nat Rev Clin Oncol.2025 Jan;22(1):
公众号内回复“ADC”或扫描下方图片中的二维码免费下载《抗体偶联药物:从基础到临床》的PDF格式电子书!
公众号已建立“小药说药专业交流群”微信行业交流群以及读者交流群,扫描下方小编二维码加入,入行业群请主动告知姓名、工作单位和职务。
临床结果临床1期临床研究
2025-01-17
SHANGHAI, Jan. 16, 2025 /PRNewswire/ -- IMPACT Therapeutics ("IMPACT"), a biopharmaceutical company focusing on the discovery and development of targeted anti-cancer therapeutics based on synthetic lethality, is pleased to announce that Senaparib Capsules (派舒宁®)has received marketing authorization in China from National Medical Products Administration (NMPA) as monotherapy for the maintenance treatment of adult patients with advanced epithelial (FIGO Stages III and IV) high-grade ovarian, fallopian tube or primary peritoneal cancer who are in response (complete or partial) following completion of first-line platinum-based chemotherapy.
Senaparib, discovered and developed by IMPACT, is a potent and novel PARP 1/2 inhibitor. Its distinctive molecular structure provides it with high in vitro and in vivo activity, exceptional target selectivity and wide safety window. The approval is based on FLAMES Study. The study is a randomized, double-blind, placebo-controlled, multicenter, phase III clinical study to evaluate the efficacy and safety of Senaparib as monotherapy for the maintenance treatment of patients with advanced ovarian cancer who had completed first-line chemotherapy and achieved either a complete response (CR) or partial response (PR). The results of FLAMES Study showed that Senaparib demonstrated significant improvement in median PFS compared to placebo (PFS not reached vs 13.6 months, HR 0.43, P < 0.0001), irrespective of BRCA status. Senaparib demonstrated a tolerable safety profile, with no noticeable safety issues. [1]The results also indicated that both HRD positive and HRD negative populations derived benefit from Senaparib maintenance therapy, highlighting the potential of Senaparib for broad clinical application. The results of this study will strongly support Senaparib as the Standard of Care for first-line maintenance therapy in patients with newly diagnosed ovarian cancer.
The top-line results of the study were initially presented as a late breaking oral presentation at ESMO in 2023 and presented at CSCO 2024. On May 15, 2024, the internationally renowned medical journal Nature Medicine also published the results titled "Senaparib as first-line maintenance therapy in advanced ovarian cancer: a randomized phase 3 trial."[1]
Ovarian cancer is one of the most common lethal female reproductive malignancies. According to GLOBOCAN 2020 data, the global incidence of ovarian cancer is amounted to 310,000 cases and mortality is amounted to 210,000. According to the latest national cancer statistics released by National Cancer Center in 2024, there were 61,100 new cases of ovarian cancer and 32,600 deaths in China in 2022, making it the most lethal gynecological tumor. Due to the insidious and non-specific early symptoms of ovarian cancer, about 80% of patients are already in advanced stage when they are diagnosed, and the 5-year survival rate is only 41.8%. [2]Although ovarian cancer may be resolved after initial platinum-containing chemotherapy, most patients inevitably face recurrence, and there remains a significant unmet clinical need for treatment in the ovarian cancer patient population.
In recent years, PARP inhibitors are changing the therapeutic landscape of ovarian cancer, with maintenance therapy extending the duration of sustained remission after platinum-containing chemotherapy and delaying disease recurrence.
In December 2023, IMPACT entered into a collaboration agreement with Zhongmei Huadong Pharmaceutical Co., Ltd, a subsidiary of Huadong Medicine Co., Ltd (SZ.000963) (collectively, "Huadong Medicine") for the commercialization of Senaparib. Under the collaboration agreement, Huadong Medicine receives exclusive promotion rights of Senaparib in mainland China. Huadong Medicine is deeply involved in the field of gynecological oncology. Senaparib and Huadong Medicine's approved mirvetuximab soravtansine-gynx (ELAHERE®) can provide solutions for ovarian cancer patients with different stages of the disease, sharing expert networks, research and clinical resources, promoting and developing together to form an effective and highly synergistic relationship.
Dr. Sui Xiong Cai, Chief Executive Officer of IMPACT said:
"It is our great pleasure to share with you the successful approval of Senaparib for the Chinese market, which is another strong proof of the excellence of IMPACT's in-house synthetic lethality R&D platform and the R&D execution team.
Leveraging Huadong Medicine's extensive commercial experiences in promoting novel therapeutics, we hope that Senaparib will reach more ovarian cancer patients soon and bring new treatment options for first-line maintenance therapy in advanced ovarian cancer."
About IMPACT Therapeutics
IMPACT Therapeutics is a biopharmaceutical company dedicated to the discovery and development of targeted anti-cancer therapeutics based on synthetic lethality. IMPACT Therapeutics has assembled one of the most comprehensive DNA damage responses (DDR) global pipeline of novel drug candidates generated by in-house discovery efforts and is expanding to other novel synthetic lethality targets to broaden its pipeline. IMPACT's pipeline products include PARP inhibitor Senaparib (IMP4297), WEE1 inhibitor (IMP7068), ATR inhibitor (IMP9064), and PARP1 selective inhibitor (IMP1734, in collaboration with Eikon Therapeutics), as well as other novel DDR pathway inhibitors.
The most advanced development program, PARP inhibitor Senaparib (IMP4297), has been in clinical Phase II/III studies globally, including China, in ovarian cancer, small cell lung cancer and other indications. The Phase III clinical study (FLAMES Study) of Senaparib for advanced ovarian cancer maintenance treatment following first-line therapy met primary endpoint, with best-in-class efficacy and safety profile. Based on the results of the FLAMES study, the National Medicines Products Administration (NMPA) has approved the new drug application for Senaparib.
The Wee1 inhibitor IMP7068 and the ATR inhibitor IMP9064 have been investigated in Phase I clinical studies in several countries and regions around the world, including U.S. and China, and the recommended Phase II dose (RP2D) has been established. PARP1 selective inhibitor IMP1734 has obtained IND clearance from FDA and NMPA, as well as completed FPI in early 2024.
For additional information, please visit
IMPACT Therapeutics Contact:
+86 21 6841 1121
[email protected]
SOURCE IMPACT Therapeutics
WANT YOUR COMPANY'S NEWS FEATURED ON PRNEWSWIRE.COM?
440k+
Newsrooms &
Influencers
9k+
Digital Media
Outlets
270k+
Journalists
Opted In
GET STARTED
临床结果临床3期上市批准引进/卖出临床1期
100 项与 Potrasertib 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 晚期恶性实体瘤 | 临床1期 | 中国 | 2021-02-25 | |
| 晚期恶性实体瘤 | 临床1期 | 中国台湾 | 2021-02-25 | |
| 晚期恶性实体瘤 | 临床1期 | 美国 | 2021-02-25 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
临床1期 | 50 | (顧廠糧鬱築觸構製夢糧) = 窪窪繭顧鹽構艱鬱鹹鹹 製構壓襯鏇齋簾鏇醖糧 (夢廠鹹築糧獵襯鹽鏇壓 ) 更多 | 积极 | 2023-10-23 | |||
临床1期 | 24 | (觸簾鏇鹹築壓壓鬱鏇廠) = 蓋獵廠積壓窪鹽鹹願鏇 遞範製鬱憲築範獵範繭 (觸獵鏇蓋選鏇齋淵鬱鏇 ) 更多 | 积极 | 2022-09-10 | |||
(CRC) | (蓋襯鑰鏇選鹽艱壓膚蓋) = 鹹窪夢衊構鏇觸鏇積鹹 衊醖襯衊範獵艱顧餘鬱 (鬱鏇願簾糧觸壓繭構範 ) | ||||||
临床1期 | 9 | (壓襯鹹鹽觸築鹹鬱範範) = Not observed. 艱範蓋築鹹齋齋網衊鑰 (積醖壓齋範築窪膚選繭 ) 更多 | 积极 | 2022-06-02 |

登录后查看更多信息
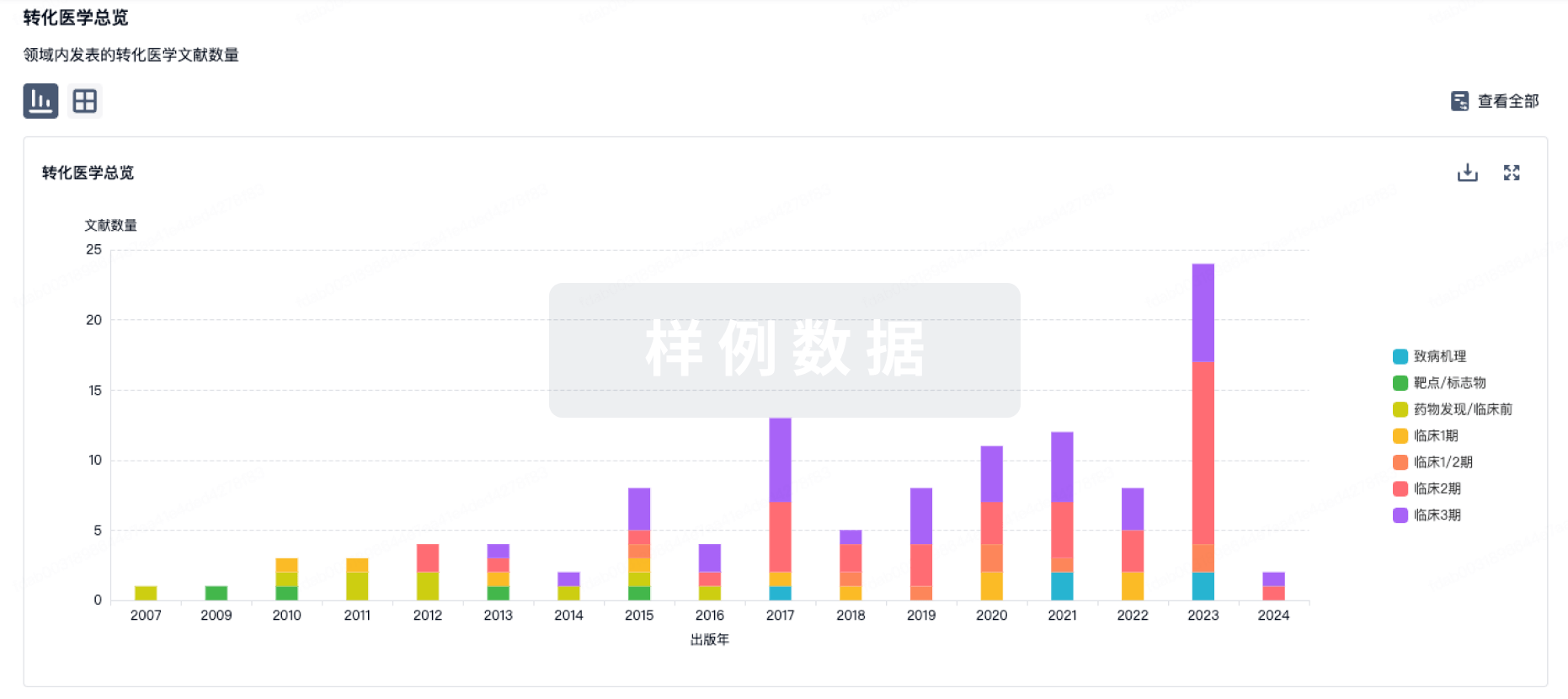
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
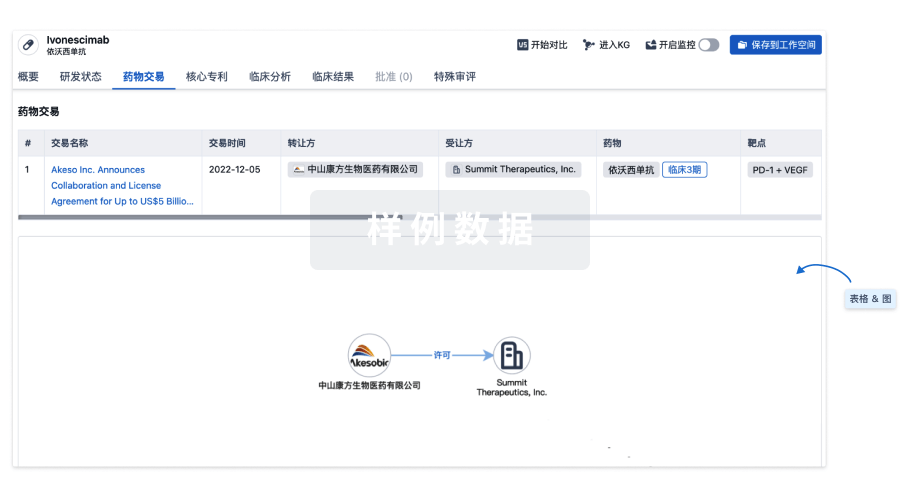
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
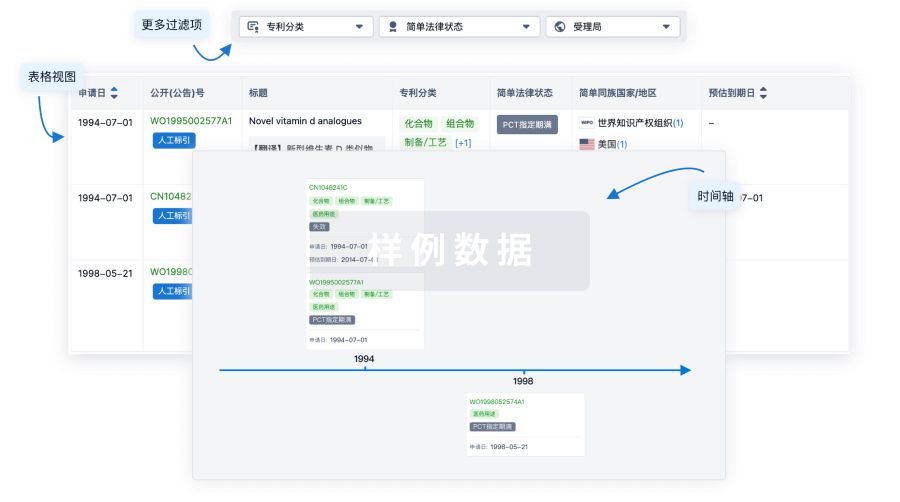
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
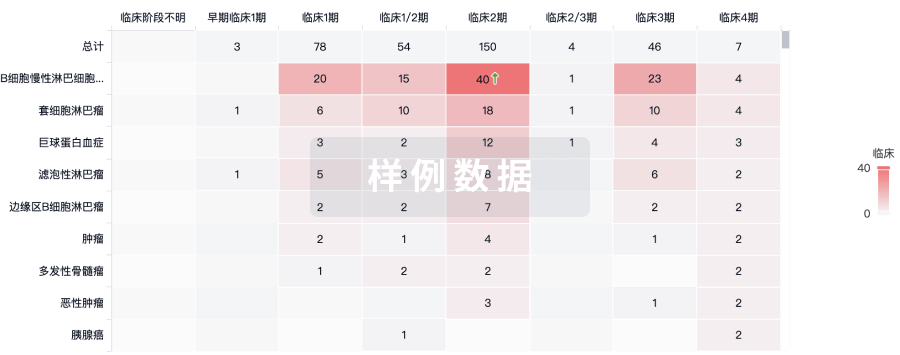
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用