预约演示
更新于:2025-08-05
RBD-7022
更新于:2025-08-05
概要
基本信息
药物类型 siRNA |
别名 QLC 7401、QLC-7401、QLC7401 + [2] |
靶点 |
作用方式 抑制剂 |
作用机制 PCSK9抑制剂(前蛋白转化酶枯草杆菌蛋白酶Kexin-9抑制剂) |
在研适应症 |
非在研适应症- |
原研机构 |
非在研机构- |
最高研发阶段临床2期 |
首次获批日期- |
最高研发阶段(中国)临床2期 |
特殊审评- |
登录后查看时间轴
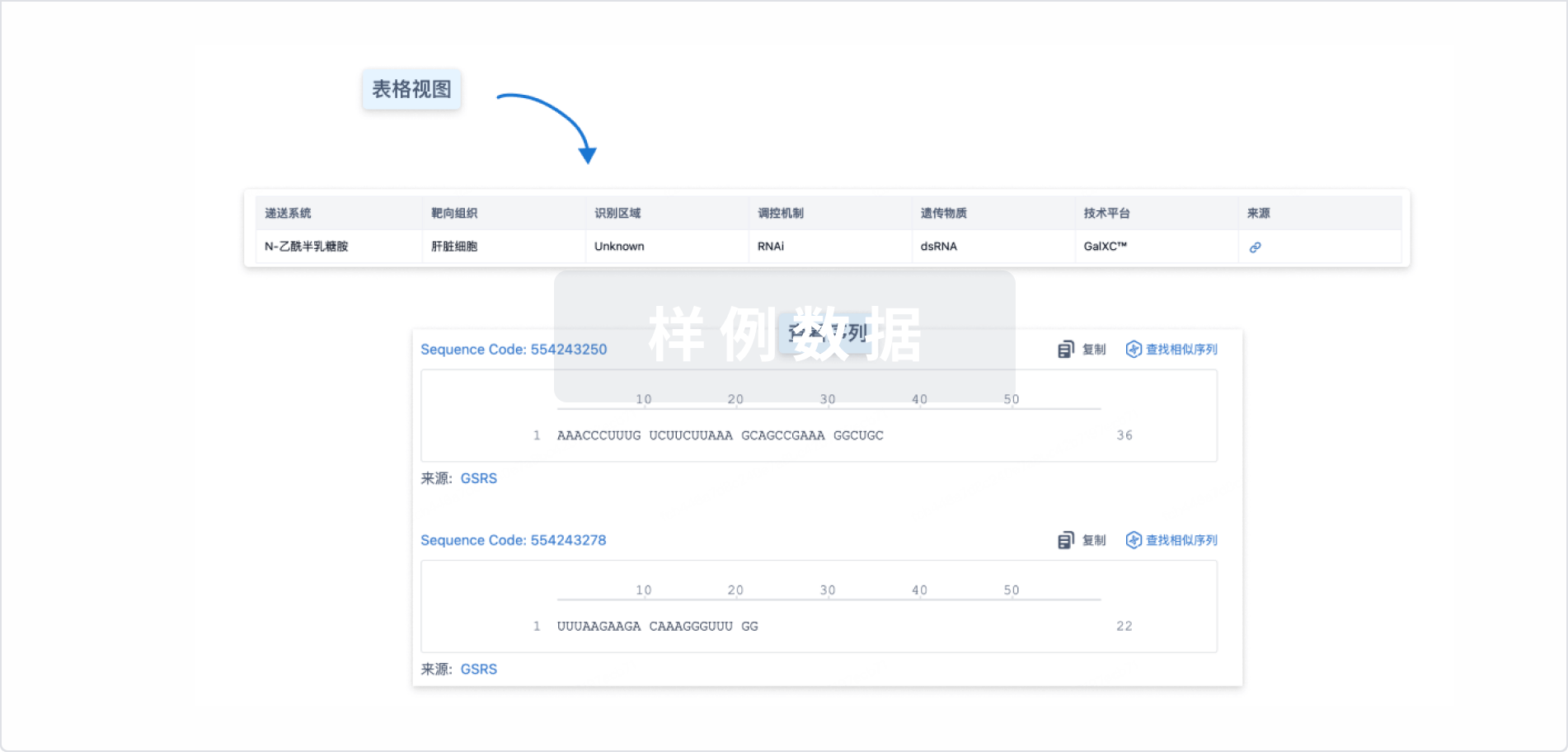
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
关联
5
项与 RBD-7022 相关的临床试验NCT07074236
A Randomized, Open-label, Single-center, Parallel Study in Subjects With Normal or Elevated Low-density Lipoprotein Cholesterol Level to Compare the Pharmacokinetics and Pharmacodynamics of the Two Formulations of QLC7401 Injection
The purpose of this study is to compare the pharmacokinetics and pharmacodynamics behavior of the two formulation of QLC7401 injection to further evaluate the effect of the production site change.
开始日期2025-07-01 |
申办/合作机构 |
NCT06750341
A Multicenter, Randomized, Double-blind, Placebo-controlled Phase II Clinical Study to Evaluate the Efficacy and Safety of Different Doses of QLC7401 in Patients With Primary Hypercholesterolemia or Mixed Hyperlipidemia With Poorly Controlled Low-density Lipoprotein Cholesterol (LDL-C) Elevated on Optimized Lipid-lowering Therapy
To evaluate the efficacy, safety, pharmacodynamics and immunogenicity of QLC7401 subcutaneous administration in patients with primary hypercholesterolemia or mixed hyperlipidemia with poorly controlled LDL-C elevated on optimized lipid-lowering therapy.
开始日期2025-02-01 |
申办/合作机构 |
CTR20244751
评价不同剂量QLC7401在接受优化降脂治疗控制不佳的低密度脂蛋白胆固醇(LDL-C)升高的原发性高胆固醇血症或混合型高脂血症患者中有效性和安全性的多中心、随机、双盲、安慰剂对照II期临床研究
主要研究目的:评价不同剂量QLC7401在接受优化降脂治疗控制不佳的LDL-C升高的原发性高胆固醇血症或混合型高脂血症受试者中的安全性、耐受性和有效性。
次要研究目的:评估QLC7401的药效动力学(PD)特征;评估QLC7401的免疫原性。
开始日期2025-01-20 |
申办/合作机构 |
100 项与 RBD-7022 相关的临床结果
登录后查看更多信息
100 项与 RBD-7022 相关的转化医学
登录后查看更多信息
100 项与 RBD-7022 相关的专利(医药)
登录后查看更多信息
42
项与 RBD-7022 相关的新闻(医药)2025-07-03
点击上方的 行舟Drug ▲ 添加关注全球小核酸药物的上市及临床研究现状分析来源《中国新药杂志》 2025年 第34卷 第12期作者杜慧,肖宇锋,张玢中国医学科学院/北京协和医学院医学信息研究所/图书馆摘要目的: 本研究旨在分析总结全球小核酸药物的上市及临床研究现状,探索其未来发展方向。方法: 利用药物早期研发情报数据库检索上市及临床在研的小核酸药物并对这些药物的分类、研发国家及机构、靶点、适应证和递送系统等信息进行详细分析。结果: 截至2024年9月18日,全球共有17个小核酸药物获批上市、192个小核酸药物处于临床研究状态,其中反义寡核苷酸药物和小干扰RNA药物数量较多。大多数临床在研的小核酸药物仍处于早期研发阶段,适应证已逐渐从早期的遗传病拓展到肿瘤、心血管疾病等慢性病领域,其中N-乙酰半乳糖胺偶联物是应用最广泛的递送系统。我国小核酸药物发展态势良好,企业管线布局以乙型肝炎和慢性病为主。结论: 小核酸药物已成为继小分子药物、抗体药物后的第三大类药物,其研发潜力巨大,市场前景广阔。未来的发展方向包括探索新的治疗靶点、多学科合作开发肝外递送系统以及加强产学研的深度合作,共同助力推进临床转化。关键词小核酸药物; 反义寡核苷酸; 小干扰RNA; 临床研究_正文_小核酸药物是由十几个到几十个核苷酸串联组成的短链核酸,通过干扰或调控基因表达实现疾病治疗目的。与传统的小分子化学药和抗体类药物相比,小核酸药物具有研发周期短、研发成功率高、候选靶点丰富、适应证分布广泛和治疗精准化等优点。小核酸药物主要包括反义寡核苷酸(antisense oligonucleotide,ASO)、小干扰RNA(small interfering RNA,siRNA)、微小RNA(microRNA,miRNA)、小激活RNA(small activating RNA,saRNA)、向导RNA(small-guide RNA,sgRNA)、适配体(aptamer)以及抗体核酸偶联药物(antibody-oligonucleotide conjugates,AOCs)、多肽核酸偶联药物(peptide-oligonu cleotide conjugates,POCs)等[1]。ASO是一类化学合成的单链寡核苷酸分子,通常由13~30个核苷酸组成,ASO通过沃森-克里克(Watson-Crick)碱基配对原则与靶标序列形成双链结构,从而影响靶基因的表达[2]。RNA干扰(RNA interference,RNAi)是将外源或内源RNA导入细胞后与靶基因的信使RNA(messenger RNA,mRNA)结合,从而阻断靶基因mRNA的翻译,进而抑制靶基因表达的一种技术。基于RNAi的小核酸药物有siRNA和miRNA,其中siRNA是一类长度为20~25个核苷酸的双链RNA分子,miRNA是内源性生成的单链RNA分子,长度通常为21~23个核苷酸[3]。saRNA是一种双链RNA分子,能够在转录水平上激活基因表达,而sgRNA则是单链RNA分子,在CRISPR/Cas9系统中起到导航作用,引导Cas9核酸酶到特定的基因位点进行编辑。适配体是一类具有特定三级结构的单链短核苷酸分子,与其他小核酸药物不同,适配体不通过碱基配对,而是类似于抗体,依靠其三维结构特异性结合配体。此外,AOCs和POCs将小核酸药物与抗体或多肽结合,提高小核酸药物的靶向性和递送效率[4]。自20世纪90年代初以来,小核酸药物领域经历了多次重大突破。1993年,Ambros和Ruvkun在秀丽隐杆线虫中首次发现miRNA,揭示了其在转录后基因调控中的重要作用[5]。1998年,Mello和Fire进一步阐明了RNAi的机制,为小核酸药物的发展奠定了理论基础,并于2006年荣获诺贝尔生理学或医学奖[6]。2023年,Karikó和Weissman因在核苷碱基修饰方面的贡献获得诺贝尔生理学或医学奖,其发现极大推动了mRNA疫苗和疗法的发展[7]。2024年,诺贝尔生理学或医学奖再次授予Ambros和Ruvkun,以表彰其在miRNA发现及miRNA在转录后基因调控中的作用方面作出的开创性贡献[8]。这些重要科学发现推动了小核酸药物的发展,国内外各大制药企业纷纷通过自主研发或合作引进管线等方式布局[9]。本研究使用科睿唯安公司的药物早期研发情报数据库(Cortellis Drug Discovery Intelligence,CDDI)检索上市及临床在研的小核酸药物,分析其分类、研发国家及机构、靶点、适应证和递送系统等信息,总结小核酸药物研发现状,探索其未来发展方向。资料与方法1 数据来源以科睿唯安公司CDDI数据库为检索来源,利用CDDI数据库检索“药物和生物制剂(Drugs&Bi ologics)”,在“药物分类(Product Category)”字段,限定“Antisense Therapy和Oligonucleotides,siRNA,mi croRNA,aptamer,saRNA,sgRNA,AOC,POC”,在“最高研发状态(Highest Phase)”字段,限定“Launched,Pre-Registered,PhaseⅢ,PhaseⅡ/Ⅲ,PhaseⅡ,PhaseⅠ/Ⅱ,PhaseⅠ”,共检索到17个已上市的小核酸药物和192个研发活跃(12~18个月内有进展的小核酸管线,去掉暂停/停止/未报告进展/非积极的项目)并处于临床研究阶段的小核酸药物,检索时间为2024年9月18日。2 数据处理数据处理阶段,对每种药物进行详细的临床试验匹配。利用CDDI数据库,针对研究范围内的192个小核酸药物,逐一访问其“临床试验(Clinical&Studies)”模块,并通过查阅和对比,实现了药物与相应临床试验的精确关联。文章内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明【来源:凡默谷公众号】。结果1 已上市药物情况截至2024年9月18日,全球共有17款小核酸药物获批上市,见表1。1998年,首款ASO药物福米韦生获美国FDA批准上市,用于治疗获得性免疫缺陷综合征患者并发的由巨细胞病毒(cytomegalov irus,CMV)引起的视网膜炎。随着高效抗逆转录病毒治疗(highly active antiretroviral therapy,HAART)的发展,CMV感染病例显著减少,导致福米韦生的市场需求和销售额大幅下滑,该药于2002年在欧洲退市[10]。TTR: 转甲状腺素蛋白, transthyretin; SOD1: 超氧化物歧化酶1, superoxide dismutase 1; DMD: 抗肌萎缩蛋白基因; APOC3: 载脂蛋白C3, apolipoprotein C3; SMN2: 运动神经元存活基因2, survival of motor neuron 2; LDHA: 乳酸脱氢酶A, lactate dehydrogenase A; PCSK9: 前蛋白转化酶枯草溶菌素9, proprotein convertase subtilisin /kexin type 9; HAO1: 羟基酸氧化酶1, hydroxyacid oxidase 1; ALAS1: 5-氨基乙酰丙酸合酶1,5 -aminolevulinate synthase 1; LNP: 脂质纳米颗粒, lipid nanoparticle; PS: 硫代磷酸酯; 2'-O-MOE: 2'-O-甲氧基乙基; 2'-O-Me: 2'-O-甲氧基; 2'-F: 2'-氟; 5mC: 5-甲基胞苷; PMO: 磷酰二氨基吗啉代寡核苷酸, phosphorodiamidate morpholino oligomer2006年,RNAi机制获得诺贝尔生理学或医学奖,极大推动了该领域的研究与开发。然而,RNAi的不稳定性、潜在的免疫原性及递送系统的缺乏导致研发进展缓慢,大型制药企业削减投资,行业发展陷入停滞。2014年,N-乙酰半乳糖胺偶联物(N-acetylgalactosamine conjugate,GalNAc)作为一种小核酸偶合递送系统飞速发展,促进了小核酸药物研发的迅速复苏[11]。随着化学修饰和递送系统技术的进步,小核酸药物进入了稳健且快速发展的时代。2016年后多款重磅小核酸药物陆续上市,见图1。仅2023年全球共批准了4款小核酸药物,包括2款ASO药物、1款siRNA药物和1款适配体药物。全球17款获批小核酸药物中ASO药物数量最多(共10款),其次为6款siRNA药物和1款适配体药物。国家药品监督管理局(National Medical Products Administration,NMPA)共批准了2款ASO药物和1款siRNA药物,分别是tofersen(2024年)、nusin-ersen(2019年)和inclisiran(2023年)。图1 2016—2023 年全球获批的小核酸药物数量分布遗传性疾病是目前获批最多的适应证类别,17种上市的小核酸药物中,有11种针对遗传性疾病(遗传性转甲状腺蛋白淀粉样变性、杜氏肌营养不良症、家族性乳糜微粒血症综合征、脊髓性肌萎缩、急性肝卟啉症)、1种用于眼科疾病(年龄相关性黄斑变性引起的地图样萎缩)、1种用于神经退行性疾病(肌萎缩侧索硬化症)、1种针对心血管疾病(高胆固醇血症)、1种用于血液肿瘤(骨髓增生异常综合征)、2种用于代谢疾病(1型原发性高草酸尿症)。2021年inclisiran作为饮食的辅助疗法获批,用于成人原发性高胆固醇血症或混合型血脂异常患者的治疗,该药适用于在接受最大耐受剂量的他汀类药物治疗仍无法达到低密度脂蛋白胆固醇(low-density lipoprotein cholesterol,LDL-C)目标水平的患者。inclisiran的获批标志着小核酸药物开始在慢性病管理中展现其独特的优势和潜力[12]。未经修饰的小核酸药物稳定性差且特异性低,容易引起脱靶毒性。化学修饰可以增强小核酸药物对核酸酶的抗性,减少免疫原性和脱靶效应,同时提高靶向性[13]。常见的化学修饰涉及对磷酸骨架、核糖和碱基的修饰,包括PS,2'-O-Me,2'-O-MOE,2'-F和5mC[14]。目前上市的11种ASO药物和6种siR NA药物均采用了骨架、核糖和碱基的化学修饰。PMO呈电中性,骨架中磷酸二酯键被磷酰二氨基键取代,核糖被吗啉环取代。PMO具有生物稳定性强、带中性电荷以及不依赖核糖核酸酶H(ribonuclease H,RNase H)的作用机制等优点[15-16]。基于PMO修饰的4种ASO药物eteplirsen,golodirsen,viltolarsen,casimersen获批用于杜氏肌营养不良症的治疗。核酸药物由于相对分子质量较大、容易降解和无法自由进入细胞等缺点,难以在体内发挥作用。递送系统可以保护核酸药物免于降解,确保其稳定性,并将其有效递送到靶组织。目前上市的小核酸药物采用了2种递送系统LNP和GalNAc,见图2。GalNAc是唾液酸受体(asialoglycoprotein receptor,ASGPR)的靶向配体,可以与肝脏实质细胞表面的ASGPR特异性结合,实现细胞的快速胞吞,该技术具有高度的肝靶向特异性[17]。在6种使用递送系统的siRNA药物中,5种采用了GalNAc偶联递送系统、1种使用了LNP递送系统。eplontersen是首个获批上市的GalNAc偶联ASO药物,是Ionis公司继inotersen之后的第2种遗传性转甲状腺蛋白淀粉样变性治疗药物,相比inotersen,eplontersen的给药剂量更小、给药频率更低[18]。2 临床研究阶段的药物研发情况截至2024年9月18日,全球共有192个研发活跃并处于临床研究阶段的小核酸药物,其中ASO药物86个,是临床研发数量最多的小核酸药物,约占45%; siRNA药物也是小核酸药物领域的研究热点之一,约占35%。此外,适配体、sgRNA、miRNA及saRNA的药物研发尚处于早期阶段,数量较少,见图3。小核酸药物的研发在全球多个国家和地区均有分布,其中美国、欧洲和中国是主要的研发基地。美国在小核酸药物的研发方面处于领先地位,目前有109个小核酸药物处于临床研究状态,远超其他国家。在欧洲,英国、瑞士、荷兰和德国的小核酸药物临床研究较为活跃。我国有25个小核酸药物处于临床研究状态,其中siRNA和sgRNA研发管线布局较多,ASO药物研发参与较少,见表2。全球范围内研发小核酸药物的企业中,Ionis公司和Alnylam公司布局最为广泛。Ionis公司专注于ASO药物研发,有32个临床在研的小核酸药物。Alnylam公司是RNAi领域的领军企业,有12个临床在研的小核酸药物。此外,Biogen公司、Arrow head公司、Lilly公司也较为活跃。我国小核酸药物发展呈现良好态势,多家企业积极布局小核酸药物的研发管线,瑞博生物公司、圣因生物公司、舶望制药公司和本导基因公司等企业都在积极推进多个项目的临床试验。全球临床在研的小核酸药物仅有9%的药物最高研发状态为预注册和Ⅲ期临床试验,大多数集中在Ⅰ期和Ⅱ期临床试验阶段。处于Ⅲ期临床试验的药物中以ASO和siRNA药物为主,适配体、sgRNA、miRNA、saRNA药物大多处于早期临床研究阶段,见图4。小核酸药物因其独特的机制和广泛的治疗潜力,能够针对多种疾病中的关键基因进行精准调控。与传统药物相比,小核酸药物能够作用于更广泛的靶点。目前,全球临床在研的小核酸药物中,针对HBV和DMD的靶点研究最为集中。此外,针对心血管疾病(如载脂蛋白和AGT)以及代谢性疾病[如PCSK9和patatin样磷脂酶结构域蛋白3(patatin-like phospholipase domain containing3,PNPLA3)]靶点也有多个药物正在进行临床研究,见表3。我国小核酸药物的靶点多集中于乙型肝炎和慢性病领域,较少涉及罕见病。全球小核酸药物的适应证分布广泛,涉及遗传性疾病、肿瘤、神经系统疾病、代谢疾病、感染、心血管系统疾病等多个疾病领域,见图5。目前上市的小核酸药物大多集中在遗传性疾病领域,如杜氏肌营养不良症、脊髓性肌萎缩症和遗传性转甲状腺蛋白淀粉样变性。然而,临床在研的小核酸药物适应证已逐渐从早期的遗传病拓展到肿瘤、心血管疾病、代谢疾病等慢性病领域。在这一趋势中,我国制药企业大多将布局重心放在慢性病领域,如信达生物公司的高血压治疗药物SGB-3908、圣因生物公司的高血脂治疗药物SGB-9768、舶望制药公司的高血脂治疗药物BW-01、瑞博生物公司的高血脂治疗药物RBD-7022。此外,乙型肝炎也是我国制药企业布局较为集中的领域,包括浩博医药公司、瑞博生物公司、恒瑞医药公司、舶望制药公司、星曜坤泽公司和正大天晴公司在内的多家企业均有相关药物在研。化学修饰和递送系统的进步推动了小核酸药物的发展和临床应用。目前已上市的ASO药物大多仅采用化学修饰,未使用特殊的递送系统。eplontersen是唯一获批上市的GalNAc-ASO偶联药物。多家制药公司如Ionis公司、CiVi Biopharma公司和Novartis公司正在积极开发GalNAc-ASO偶联药物。其中Ionis公司的临床进展最为迅速,已有2款产品进入预注册阶段、1款产品进入Ⅲ期临床试验,见表4。GalNAc是目前小核酸药物应用最广泛的递送系统,但仅能靶向肝脏细胞。为了实现肝外递送目的,多项临床研究开发了新型递送系统,包括AOCs、POCs、外泌体和聚合物基质等,见表5。AOCs将小核酸药物的靶点特异性与抗体的组织特异性相结合,协同发挥了这2种分子的优势[19-20]。Avidity Biosciences公司和Dyne Therapeutics公司在AOCs研发领域表现突出,其中Avidity Biosciences公司的强直性肌营养不良药物AOC-1001已进入Ⅲ期临床试验阶段。POCs将多肽与小核酸药物偶联,显著改善了小核酸药物的递送、细胞摄取和生物利用度。细胞穿透肽(cell-penetrating peptides,CPP)是一类能够有效穿过细胞膜的短肽序列,是POCs中研究最多的偶联物之一[21-22]。进展较快的POCs已进入Ⅱ期临床试验阶段。Sarepta Therapeutics公司的vesleteplirsen是已获批上市的eteplirsen与CPP偶联而成,用于治疗杜氏肌营养不良症。PepGen公司基于其增强递送寡核苷酸(enhanced delivery oligonucleotide,EDO)技术平台设计的PGN-EDO51也是一种多肽-寡核苷酸偶联物,用于治疗杜氏肌营养不良症[23]。外泌体是内源性的纳米级细胞外囊泡,具有免疫原性低、安全性高的特点[24]。Codiak BioSciences公司研发的exoASO-STAT6已进入Ⅰ期临床试验,该药物能够选择性递送ASO以破坏肿瘤相关巨噬细胞(tumor-associated macrophages,TAM)中的STAT6信号传导并诱导抗肿瘤免疫反应[25]。siG12D LODER是一种由可生物降解的聚合物基质包裹的针对KRAS G12D突变的siRNA,临床研究(NCT01676259)表明siG12D LODER联合化疗可延长KRAS G12D/V突变局部晚期胰腺癌患者总体生存期9.3个月[26]。讨论目前获批上市和处于临床研究状态的小核酸药物中,ASO药物因上市较早和成熟的商业化发展在小核酸药物研发中的占比较高,其次为siRNA药物。在靶点和适应证方面,上市的小核酸药物主要集中在罕见病领域,临床在研项目已开始向肿瘤、糖尿病、高血脂、乙型肝炎等领域拓展,但靶点已出现同质化现象,针对HBV、载脂蛋白、PCSK9的研究较为集中。临床分期方面,大多数小核酸药物仍处于早期研发阶段,而处于Ⅲ期临床试验的药物主要以ASO和siRNA药物为主。欧洲和美国在小核酸药物研发领域仍处于领先地位,以Ionis公司和Alnylam公司为代表的大型制药企业在行业发展初期主要聚焦于致病机制明确、缺乏有效治疗手段的遗传性罕见病,通过快速验证潜在治疗靶点的有效性,借助孤儿药(orphan)、快速通道(fast track)、加速审批(accelerated approval)、优先审评(priority review)和突破性疗法(break- through therapy)等多条特殊认定和审评途径,实现了药物的快速获批和上市[27]。在化学修饰和递送系统技术成熟后,逐步向心血管疾病及代谢疾病过渡。相比之下,我国对罕见病的研究相对较少,而更多集中在乙型肝炎和高血压、高血脂等慢性病领域,这可能与国内罕见病政策体系有待完善和罕见病药物研发成本较高有关[28]。尽管我国在小核酸药物研发方面起步较晚,但在政策扶持和科研投资增加的推动下,国内多家企业正积极布局临床开发。2024年瑞博生物公司和舶望制药公司先后完成了License-out授权,将企业的专利技术、药物候选物或研发平台授权给国际制药企业[29-30],展现了国内制药企业在全球小核酸药物研发领域中的活跃参与和贡献,再次证明了我国核酸药物发展的良好态势。未来随着国内小核酸药物研发能力的不断提升,有望逐步进入突破型创新阶段。核酸药物具有其他药物所不具备的独特优势,但也面临诸多挑战。其在体内的稳定性较差、难以穿透细胞膜、靶向性不足,因而递送系统的开发一直是研发过程中需要解决的关键难题[31]。目前,Gal-NAc-siRNA递送系统已较为成熟,已上市的6种siRNA药物中有5种采用了GalNAc系统。但上市的ASO药物大多数仅采用了结构修饰,虽然在一定程度上解决了直接递送的难题,但大剂量给药可能会限制其临床应用以及增加用药风险。首款Gal NAc-ASO药物eplontersen的获批以及多项GalNAc ASO药物临床试验的推进标志着ASO药物开始进入“GalNAc递送系统”时代。GalNAc递送系统具有高效的肝脏靶向性,在治疗肝脏疾病方面表现出显著优势,但要实现对肝外组织/细胞的靶向递送并达到更安全、更稳定和更高效的递送效果,仍然面临巨大挑战。目前多项临床试验对肝外递送系统进行了研究,包括开发AOCs、POCs、外泌体、聚合物基质等,但同时也面临提高递送效率、降低毒性和免疫原性、减少研发成本等问题[4]。小核酸药物作为一种新型治疗方式,在罕见病和慢性病治疗领域已展现出显著潜力。为了充分释放其治疗潜力,需要克服一系列挑战,探索新的治疗靶点和拓展适应证、避免靶点同质化对于推动小核酸药物的发展至关重要。同时需要来自物理、生物、化学和工程学等多学科的科研人员共同努力,推动肝外递送系统的发展,以解决当前递送技术的局限性。此外,加强产学研的深度合作、促进基础研究与临床应用的紧密结合、加快药物的临床转化是实现小核酸药物广泛应用的重要途径[32]。未来,小核酸药物有望成为具有更大应用潜力的治疗手段,进一步丰富治疗药物的选择,为患者的健康和福祉作出更大的贡献。参考文献详见《中国新药杂志》 2025年 第34卷 第12期文章信息源于公众号凡默谷,登载该文章目的为更广泛的传递行业信息,不代表赞同其观点或对其真实性负责。文章版权归原作者及原出处所有,文章内容仅供参考。本网拥有对此声明的最终解释权,若无意侵犯版权,请联系小编删除。学如逆水行舟,不进则退;心似平原走马,易放难收。行舟Drug每日更新 欢迎订阅+医药大数据|行业动态|政策解读
寡核苷酸信使RNA核酸药物siRNA上市批准
2025-05-27
·同写意
5月26日,信立泰宣布与国为生物达成协议,获得其自主研发的AGT-siRNA药物GW906在中国市场的独家许可权益。根据协议,国为医药将获得最高1.8亿元的首付款及研发里程碑款总金额。未来产品获批上市后,销售里程碑累计最高达3.7亿元。同时,在协议约定期限内,国为医药将获得年度净销售额按一定比例支付销售提成。不只是上述两家公司,近期,包括礼来、艾伯维在内的MNC都相继披露了siRNA的研发动向。自1998年RNAi现象被揭示,到2020年全球首个长效降脂siRNA药物Inclisiran横空出世,这条被科学界称为“基因沉默革命”的技术赛道,历经多次发展瓶颈。如今,又到新的爆发期。TONACEA01长效优势GW906是国为医药基于siRNA技术平台开发的一款靶向血管紧张素原(AGT)的siRNA药物,拟开发的适应症为原发性高血压,目前正在开展I期临床研究。AGT是肾素-血管紧张素-醛固酮系统(RAAS)的最上游前体蛋白,通过抑制其合成可长效降低血压。GW906通过靶向肝脏中的AGT mRNA,抑制AGT的表达,减少血管紧张素II生成,从而舒张血管、降低血压。与传统的高血压治疗药物相比,GW906靶向AGT这一RAAS系统最上游前体物质,为解决长期口服RAAS阻滞剂(如ACEI和ARB)的醛固酮逃逸问题提供了潜在的可能性;同时,相比较ACEI类药物,AGT-siRNA药物不会导致缓激肽蓄积,从而有可能避免咳嗽、血管性水肿、呼吸困难和疼痛等不良反应。 GW906通过间歇性皮下注射,以期实现长期、稳定的降压效果,进而提高患者依从性。临床前数据显示,GW906的疗效和安全性良好,具备长效的潜力。高血压是中国患者基数最大的慢性病之一,患病人数超3亿,但治疗率与控制率不足50%,存在巨大的未满足需求。全球范围内,针对AGT的siRNA药物研发尚处于早期阶段。GW906若成功上市,有望凭借长效优势抢占细分市场。国为医药之外,也有不少国内药企布局AGT siRNA药物,包括舶望制药BW-00163、石药集团SYH2062、信达生物与圣因生物合作开发的IBI3016(SGB-3908)、成都先导LDR2402,均已进入临床研究阶段。图源:太平洋证券研报TONACEA02基因沉默革命2006年,诺贝尔生理学或医学奖授予RNAi现象的发现者,正式将siRNA疗法推向生物医药舞台中央。然而,早期研究遭遇致命瓶颈:传统LNP递送系统难以精准靶向组织,药物稳定性差、生产成本高企,导致辉瑞、罗氏等跨国药企相继缩减管线,行业一度陷入技术冰河期。转机出现在2010年,随着GalNAc偶联技术横空出世,siRNA药物开发迎来转折点。这项递送系统通过与肝细胞表面受体特异性结合,将药物精准导航至靶器官,将给药剂量降低至毫克级。与此同时,化学修饰技术大幅延长药物半衰期,工业化生产体系突破量产瓶颈。2018年,Alnylam研发的Patisiran获得FDA批准,成为全球首个治疗遗传性转甲状腺素蛋白淀粉样变的siRNA药物,标志着该技术正式跨越“死亡之谷”。2020年诺华推出的Inclisiran更以“半年一针”的超长效特性,在心血管领域掀起治疗模式革命,销售额4年内暴涨近63倍,成功验证siRNA疗法在慢性病领域的治疗潜力。目前,全球已有近10款siRNA药物获批上市。据Evaluate Pharma数据,全球siRNA药物市场规模已突破50亿美元,预计2030年将达200亿美元。近期,不少MNC披露有关siRNA药物的研发动向。5月,艾伯维宣布以3.35亿美元首付款,以及潜在数十亿美元与ADARx Pharmaceuticals达成协议,开发适用于多个疾病领域的siRNA新药,包括神经科学、免疫学和肿瘤学。3月底,礼来宣布其新型siRNA药物lepodisiran在II期临床试验中取得积极结果:在最高剂量下可将Lp(a)水平平均降低93.9%,远超现有降脂手段,且单次给药效果可持续近一年。赛诺菲和Alnylam联合开发的潜在重磅siRNA疗法Qfitlia(fitusiran),也已在3月获FDA批准。这款疗法通过抑制肝脏中抗凝血酶的生产,有望成为一种革命性的预防手段,造福所有血友病患者。众多国内药企同样布局了丰富的siRNA药物管线,适应证主要集中在高脂血症、高血压、乙肝,靶点覆盖PCSK9、ANGPTL3、AGT、COX-2等。据不完全统计,约10款国产长效siRNA制剂进入临床研究阶段,包括齐鲁制药从瑞博生物引进的RBD7022、圣因生物SGB-3403、石药集团SYH2053、君实生物RP910、悦康药业YKYY015等,大多处于Ⅰ期临床。另外,国内药企在这一领域成功与MNC牵手。石药集团在2024年10月与阿斯利康达成总金额为20.2亿美元的交易,授权后者临床前阶段的Lp(a)抑制剂YS2302018;默沙东在今年3月则花费近20亿美元,引进恒瑞医药处于II期临床阶段的Lp(a)抑制剂HRS-5346。随着全球siRNA技术进入爆发期,期待这款疗法能在更广的市场中开辟新天地。参考文章:1、信立泰公告:获AGT-siRNA药物GW906独家许可权益2、siRNA药物,已在爆发前夜;新浪医药关于同写意 同写意论坛是中国新药研发行业权威的多元化交流平台,二十一年来共举办会议论坛百余期。“同写意新药英才俱乐部”基于同写意论坛而成立,早已成为众多新药英才的精神家园和中国新药思想的重要发源地之一。同写意在北京、苏州、深圳、成都设立多个管理中心负责同写意活动的运营。尊享多重企业/机构会员特权 ● 分享庞大新药生态圈资源库;● 同写意活动优享折扣;● 会员专属坐席及专家交流机会;● 同写意活动优先赞助权;● 机构品牌活动策划与全方位推广;● 秘书处一对一贴心服务。入会请联系同写意秘书处 同写意创新链盟机构 (上下滑动查看更多)深势科技 | 新天地药业 | 快舒尔医疗 | 华赛伯曼 | 艾里奥斯 | 药明合联 | 皓元医药 | 希格生科 | 纽瑞特医疗 | 夸克医药 | 石药集团 | 源生生物 | 君赛生物 | 达尔文生物 | 浩博工程 | 怀雅特 | 赛立维 | 科伦博泰 | 赛隽生物 | 安升达/金唯智 | 卡替医疗 | 达科为生物 | 沙利文 | 天广实 | 拜耳 | 楚天科技 | 三生制药 | 三启生物 | 国通新药 | 通瑞生物 | 科济药业丨立迪生物 | 森西赛智 | 汇芯生物 | 申科生物 | 方拓生物 | 东抗生物 | 科盛达 | 依利特 | 翊曼生物丨锐拓生物丨复百澳生物丨圆因生物丨普洛斯丨华润三九丨皓阳生物丨人福医药丨广生堂药业丨澳宗生物丨妙顺生物 | 荣捷生物丨行诚生物 | 宜联生物 | 生命资本 | 恒诺康丨斯丹姆 | 益诺思 | 深圳细胞谷丨佰诺达生物 | 沃臻生物 | 金仪盛世 | 朗信生物 | 亦笙科技 | 中健云康 | 九州通 | 劲帆医药 | 沙砾生物 | 裕策生物 | 同立海源 | 药明生基 | 奥浦迈 | 原启生物 | 百力司康 | 宁丹新药 | 上海细胞治疗集团 | 滨会生物 | FTA | 派真生物 | 希济生物 | 优睿赛思 | 血霁生物 | 优睿生物 | 邦耀生物 | 华大基因 | 银诺生物 | 百林科医药 | 纳微科技 | 可瑞生物 | 夏尔巴生物 | 金斯瑞蓬勃生物 | 健元医药 | 星眸生物 | 格兰科医药 | 莱羡科学仪器 | 明度智云 | 玮驰仪器 | 康源久远 | 易慕峰 | 茂行生物 | 济民可信 | 欣协生物 | 泰楚生物 | 泰澧生物 | 谱新生物 | 思鹏生物 | 领诺医药 | 宜明生物 | 爱科瑞思 | 阿思科力 | 博格隆生物 | 百吉生物 | 迈邦生物 | 多宁生物 | 万邦医药 | ASCT | 为度生物 | 比邻星创投 | 赛桥生物 | 吉美瑞生 | 荣泽生物 | 科金生物 | 汉超医药 | 康日百奥 | 汉腾生物 | 力品药业 | 安必生 | 博瑞策生物 | 中盛溯源 | 深研生物 | 东方略 | 赛赋医药 | 克睿基因 | 安润医药 | 镁伽科技 | 科锐迈德 | 和元生物 | 申基生物 |楷拓生物| 森松生命科技 | 凯理斯 | 尚德药缘 | 晟国医药 | 健新原力 | 纽福斯 | 华东医药 | 士泽生物 | 影研医疗科技 | 新格元生物 | 依生生物 | 腾迈医药 | 汉欣医药 | 恒驭生物 | 盛诺基 | 序祯达生物 | 乐纯生物 | 速石科技 | 耀海生物 | 新合生物 | 华龛生物 | 恺佧生物 | 成都凡微析 | 正帆科技 | 大橡科技 | 博雅辑因 | 因美纳 | 博雅控股集团 | 近岸蛋白 | 依科赛生物 | 利穗科技 | 东南科仪 | 倍谙基 | 辉诺医药 | 圣诺制药 | 埃格林医药 | 科镁信 | 爱思益普 | 复星医药 | 齐鲁制药 | 捷思英达丨荣昌生物丨泽璟制药丨奕安济世丨礼新医药丨维立志博丨派格生物丨赛生药业丨呈源生物丨启德医药丨双运生物丨宝船生物丨曙方医药丨澳斯康生物丨普莱医药丨维健医药丨海昶生物丨征祥医药丨智核生物丨望石智慧丨博生吉医药丨南京诺丹丨四星玻璃丨艾米能斯丨霁因生物丨普瑞康生物丨映恩生物丨康哲生物丨霍德生物丨海慈药业丨沃生生物丨睿健医药丨矩阵元丨斯微生物丨则正医药丨预立创投丨东立创新丨博安生物丨伟德杰生物丨星奕昂生物丨耀乘健康科技丨琅钰集团丨康德弘翼 | 原力生命科学丨上海科洲丨特瑞思丨药源丨健艾仕生物丨冠科美博丨微境生物丨天境生物丨合源生物丨泛生子丨创胜集团丨加科思药业丨丹诺医药丨凌科药业丨偶领生物丨凯斯艾生物丨成都圣诺丨松禾资本丨清普生物丨和其瑞丨开拓药业丨科兴制药丨玉森新药丨水木未来丨分享投资丨植德律所丨奥来恩丨乐明药业丨东曜药业丨君圣泰丨海创药业丨天汇资本丨再鼎医药丨济煜医药丨百英生物丨基石药业丨君实生物丨Sirnaomics,Inc.丨亦诺微丨博腾股份丨思路迪诊断丨艾博生物丨普瑞金生物丨未知君生物丨尚健生物丨阿诺医药丨有临医药丨赛业生物丨睿智医药丨博济医药丨晶泰科技丨药明康德丨创志科技丨奥星集团丨苏雅医药丨科贝源丨合全药业丨以岭药业丨科睿唯安丨DRG丨博瑞医药丨丽珠医药丨信立泰药业丨步长制药丨华素制药丨众生药业丨上海医药丨高博医疗集团丨药渡丨君联资本丨集萃药康丨诺思格丨精鼎医药丨百利药业丨Pfizer CentreOne丨默克中国创新中心丨奥来恩丨瑞博生物丨新通药物丨广东中润丨医普科诺丨诺唯赞丨康利华丨国信医药丨昆翎丨博纳西亚丨缔脉丨一品红丨和泽医药丨博志研新丨凯莱英医药丨汉佛莱丨英派药业丨京卫制药丨海思科药业丨宏韧医药丨开心生活科技丨哈三联丨Premier Research丨宣泰医药丨先声药业丨海金格丨普瑞盛医药丨Informa丨科特勒丨谋思医药丨HLT丨莱佛士丨辉瑞丨科林利康丨冠科生物丨科文斯丨卫信康丨龙沙(Lonza)丨美迪西丨阳光诺和丨润东医药丨勃林格殷格翰(中国)丨艾苏莱生物丨领晟医疗丨驯鹿医疗丨燃石医学丨中肽生化丨鸿运华宁丨泰格医药丨易迪希丨希麦迪丨百奥赛图丨迪纳利丨青云瑞晶丨鼎丰生科资本丨中源协和丨维亚生物丨青松医药丨中科谱研丨长风药业丨艾欣达伟丨鼎康生物丨中晟全肽丨海步医药丨勤浩医药丨奥萨医药丨太美医疗科技丨生特瑞丨东富龙丨Cytiva丨优辰实验室丨苏桥生物丨君达合创丨澎立生物丨南京澳健丨南京科默丨东阳光丨亚盛医药丨杰克森实验室丨上海科州丨三优生物丨三迭纪丨泰诺麦博丨Cell Signaling Technology丨PPC佳生丨澳斯康丨先为达丨智享生物丨锐得麦丨宜明昂科丨明济生物丨英百瑞丨六合宁远丨天津天诚丨百拓生物丨星药科技丨亓上生物丨真实生物丨引光医药丨方达医药丨高博医疗集团丨赞荣医药丨国投创新丨药明生物丨康哲药业丨高特佳投资丨普瑞基准丨臻格生物丨微谱医药丨和玉资本 | 倚锋资本
siRNA临床1期临床申请引进/卖出
2025-05-26
·求实药社
来源:药讯随说5月23日,苏州瑞博生物宣布其和齐鲁制药合作开发的靶向PCSK9小核酸药物RBD7022/QLC7401高脂血症2期临床(CTR20244751)完成全部患者入组。据公开信息,RBD7022是瑞博生物基于自主知识产权平台RiboGalSTARTM开发的GalNAc-siRNA药物,通过精准抑制肝脏PCSK9蛋白表达,长效降低低密度脂蛋白胆固醇(LDL-C)水平。该技术突破传统药物需频繁给药的局限,单次注射即可实现数月持续降脂,为高脂血症患者提供更优治疗选择。2023年12月,瑞博生物和齐鲁制药达成协议,将RBD7022大中华区(大陆、香港、澳门)开发、生产和商业化权利授权给齐鲁制药,而瑞博生物将获得总计超7亿人民币的首付款和里程碑付款。免责声明:文章内容仅供参考,不构成投资建议。投资者据此操作,风险自担, 关于对文中陈述、观点判断保持中立,不对所包含内容的准确性、可靠性或完整性提供任何明示或暗示的保证。请读者仅作参考,并请自行承担全部。联系我们IND 2025展位火热销售中!扫码立即咨询电话:13816031174(同微信)赞助形式包括但不仅限于演讲席位、会场展位、会刊彩页、晚宴赞助、会议用品宣传等。点击此处“阅读全文”咨询更多精彩!
siRNA临床申请引进/卖出临床2期临床1期
100 项与 RBD-7022 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 家族性混合型高脂血症 | 临床2期 | 中国 | 2025-01-20 | |
| 原发性高胆固醇血症 | 临床2期 | 中国 | 2025-01-20 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或
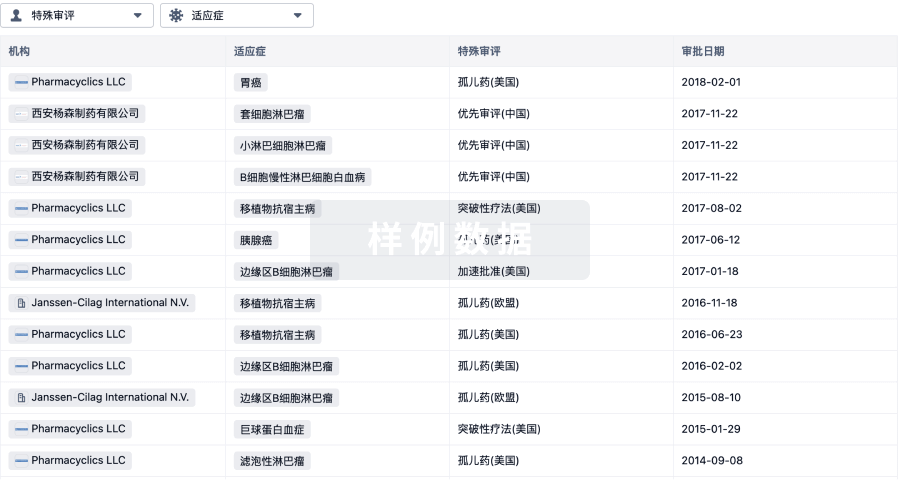
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用