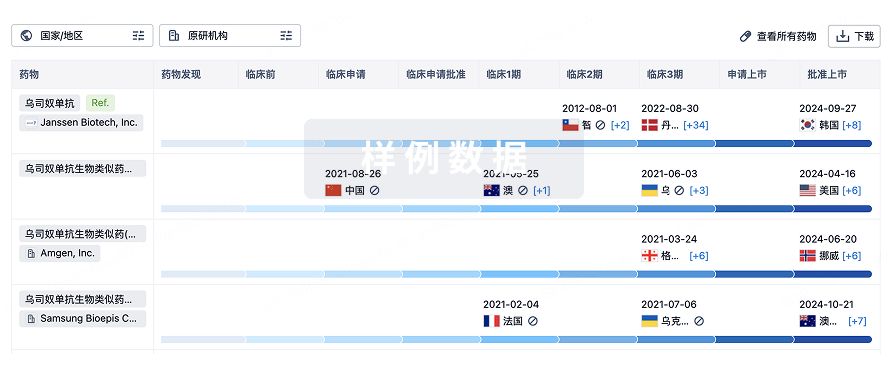
2
项与 利拉鲁肽生物类似药(珠海联邦制药) 相关的临床试验比较联邦制药利拉鲁肽注射液和诺和力治疗2型糖尿病的有效性和安全性
研究珠海联邦制药股份有限公司研制的利拉鲁肽注射液对2型糖尿病患者的有效性及安全性。
利拉鲁肽注射液在中国健康受试者中药动学和安全性比对试验
考察利拉鲁肽注射液的药动学特征,并比较中国健康成年受试者单次皮下注射利拉鲁肽注射液(受试制剂)与原研产品(参比制剂)的药动学相似性及安全性。
100 项与 利拉鲁肽生物类似药(珠海联邦制药) 相关的临床结果
100 项与 利拉鲁肽生物类似药(珠海联邦制药) 相关的转化医学
100 项与 利拉鲁肽生物类似药(珠海联邦制药) 相关的专利(医药)
20
项与 利拉鲁肽生物类似药(珠海联邦制药) 相关的新闻(医药)关于生物制品的分段生产,此前已经陆续讨论了五次。今年上半年,终于迎来了一个重大里程碑。
【案例讨论】首家ADC分段生产获批试点
【案例讨论】生物制品分段生产试点,首个产品通过检查
亦喜亦忧!再议分段生产
上海试点生物制品分段生产!花落谁家?
进口药境外生产转境内,利好CDMO几何?
5月15日国家药监局公告:通过优先审评审批程序批准北海康成(上海)生物科技有限公司申报的注射用维拉苷酶β(商品名:戈芮宁)上市,适用于12岁及以上青少年和成人I型和Ⅲ型戈谢病患者的长期酶替代治疗。
戈谢病是一种因溶酶体中葡萄糖脑苷脂酶功能缺陷导致的常染色体隐性遗传代谢病,被纳入《第一批罕见病目录》。注射用维拉苷酶β能减少葡萄糖脑苷脂在体内的贮积,从而发挥治疗作用。
同日,上海市药监局就此也有公告:该药获益于上海率先推进的生物制品分段生产试点改革,北海康成(上海)生物科技有限公司作为上市许可持有人,原液委托上海药明生物技术有限公司生产,制剂委托无锡药明生物技术股份有限公司生产。
不是因为美国法案,国内biotech脱离药明生物
上海药品审评核查中心、江苏省药监局审核查验中心在国家药监局核查中心的统筹协调下,通过串联检查等方式使得各阶段的生产和检验数据能够无缝追溯,实现全链条生产质量管理中信息的共享和互通,加快了罕见病创新生物药上市进程。
由此可见,5月15日国家药监局批准注射用维拉苷酶β上市,实现多方共赢。无论是企业还是药监部门,不仅仅是多了一个药物获批上市,更是为生物制品分段生产试点提供了实际案例。对于医生和患者来说,当然是有了新的治疗选择。
作为首个通过生物制品分段生产检查的创新生物药,注射用维拉苷酶β(研发代号CAN103)的获批无疑在国内药品监管进程中写下浓墨重彩的一笔。国家药监局和上海市药监局两级主管机构都专门发文介绍,足以见得其重大意义。
虽然首个通过生物制品分段生产检查获批的药品是落在珠海联邦的利拉鲁肽,不过,维拉苷酶β的获批在药品监管进程上显然更具有突破性。
首先,在珠海联邦的案例中,持证人和生产商是属于同一个集团,长期以来都在同一质量体系之中,管理难度相对较低,风险自然也就更低;而此次北海康成的案例,持证人和生产商是相对独立的,尽管仔细深究的话,其背后也存在资本上的关联性(2024年底药明康德持有北海康成9.50%股份),但从内部管理上来说都是独立个体。
其次,在珠海联邦的案例中,持证人和生产商都在广东药监部门的管辖范围内,且分段生产的两个场地相聚只有百米(安基路2380号和安基路2428号),如果珠海联邦向药监部门去争取视作同一场地,或许也都是可以商议的;而北海康成的案例中,分段生产的两个场地则是相隔180公里,分属于上海和江苏两地省局管辖,在监管层面的多方协调也是一次重大实践;据上海药监局公告:国家药监局药品审评核查长三角分中心与上海市药监局组建专门服务团队,在检验、审评、核查、生产许可等注册上市的全环节持续跟踪辅导,并联合市相关部门多措并举、精准发力,共同支持企业创新研发,让企业在申报注册上市过程中少走“弯路”。
最后,利拉鲁肽是一个生物类似药,此前的原研产品以及后来的多家生物类似药都已经让监管部门对其基本性质是十分熟悉的;而北海康成的注射用维拉苷酶β则是一个新分子,虽然相较于武田的注射用维拉苷酶α可能就只是糖基化存在差异,但毕竟其人体用药实践以及生产工艺和质量控制经验都是十分有限的,也就增加了监管难度。
不过,对于北海康成来说,虽然在药政上取得突破性进展并实现产品获批,自然是一片喜悦。然而在短暂喜悦过后,北海康成面对羸弱的财务状况却不敢有所懈怠,前路依然面临重重挑战。首要面对的可能就是,产品获批必然触发一笔里程碑付款,但钱从哪里来?
根据北海康成2024年报,综合现金流量表所示现金及现金等价物在2024年底还有1003万元人民币。这点钱大概率是不足以向药明生物支付这笔里程碑款项的,可能也就是够给高管发工资,2024年董事及最高行政人员薪酬为840万元,而雇员福利开支则是8905万元。截至2024年12月31日,北海康成已经将员工人数精简至67名全职员工,并且截至2025年3月中旬,进一步精简人员架构至50名全职雇员,以减少运营成本。
改善财务状况的两条路径,除了节流之外,最重要的当然是开源。
2024年收入8510万元,相比2023年达到最高峰的1.03亿元是减少的。抛开奈拉替尼在香港的分销于2023年底结束的因素之外,仍然是较2023年减少6.4%。或许,仅凭艾度硫酸酯酶β和氯馬昔巴特这两款产品销售,难以在短期内大幅度提升收入。更何况这两款产品仅有大中华区权益。
或许也可以继续向银行申请授信贷款。不够截止2024年底,公司的银行贷款和其他借款为3000万元人民币。还能从银行拿到多少钱帮助度过难关?
或许还真的只能指望刚刚获批的注射用维拉苷酶β了。有新产品可以销售,也就有了新的收入来源;参考武田收购夏尔(Shire)而获得类似品种维拉苷酶α,在过去几年的收入长期稳定在4亿美元;此外,北海康成拥有这款产品的全球权益,如果将海外权益找了合适的买家,或许能极大缓解资金压力。
作为罕见病药物第一股,北海康成所作出的各种努力对于全社会的意义,显然不言而喻。面对罕见病药物高昂的价格,支付体系的建立和完善仍然在艰难前行。无论是商业保险还是各地惠民保,都在通过全社会努力极力减轻患者家庭的负担,同时平衡药企持续发展的基本需求。
作为一家商业公司,赚钱是其基本使命,但北海康成所面临的最大尴尬是,可能无法坚持到赚钱的那一天。好在依靠分段生产试点工作,已经续命到今天。而药政突破所取得的产品成功注册,能否换来商业上的成功,是不是到头来又是做了一件公益呢?
近期活动
9月26-29日,BiG将召开以「创新出海 云起潮升」为主题的第十一届IMPACT年会,届时上海杭州双城联动,向全球化,再出发!
▼
时间及地点:
9月26-27日 上海-张江希尔顿;规模:600-800人
9月29日 杭州-西湖大学;规模:100人(邀请制,不对外开放)
报名入口
会议门票:审核制(免费);含主论坛+分论坛,不含餐;优先Biotech/Pharma、临床、院校科学家、投资机构
付费门票:详见上二维码,注册内信息
1v1资本对接会
路演项目征集
可扫码填写项目信息
现场开放1v1咨询台,与各大头部VC面对面
▼
电脑端链接:https://event.mymova.com/spa4/#/?eventname=11bigvc
共建Biomedical创新生态圈!
如何加入BiG会员?
精彩内容近日,CDE官网显示,东阳光药申报的1类新药HEC-007注射液获得临床试验默示许可,包括2型糖尿病、超重或肥胖两项适应症。消化系统及代谢疾病是东阳光药重点布局的领域之一,聚焦糖尿病、肥胖、非酒精性脂肪性肝炎(NASH)等。米内网数据显示,近年来中国三大终端六大市场(统计范围详见本文末)消化系统及代谢药(化药+生物药)销售额均超过2100亿元。据悉,HEC-007注射液是一种脂肪酸侧链修饰胰高血糖素样肽(GLP-1)/胰高血糖素(GCG)/葡萄糖依赖性促胰岛素多肽(GIP)三靶点多肽新药,通过协同激活多个受体,进一步强化减重并改善血糖血脂等代谢指标。目前国内外尚无同类产品获批上市。全球在研新药中,礼来的retatrutide处于III期临床,联邦制药的UBT-251、韩美制药的HM-15211等处于II期临床。米内网数据显示,近年来中国三大终端六大市场消化系统及代谢药(化药+生物药)销售额均超过2100亿元,但受集采等影响,其市场规模呈逐年下滑态势。近年来中国三大终端六大市场消化系统及代谢药(化药+生物药)销售情况(单位:万元)来源:米内网格局数据库消化系统及代谢药涵盖14个治疗亚类,糖尿病用药、治疗与胃酸分泌相关疾病的药物、维生素类依次位列前三,2024年市场份额分别超过31%、14%、12%。在消化及代谢疾病领域,东阳光药有14款新药在国内处于获批临床及以上阶段(不含已上市品种),重点布局糖尿病、肥胖、NASH等细分病种,覆盖全系列胰岛素(速效、短效、长效及预混)、SGLT2抑制剂、GLP-1多系列药物等,满足多样化治疗需求。东阳光药国内在研的消化系统及代谢药新药来源:米内网综合数据库胰岛素步入收获期,5款已获批上市。此外,德谷胰岛素注射液提交NDA,原研产品2024年在中国三大终端六大市场销售额接近9亿元;德谷门冬双胰岛素注射液步入III期临床,原研产品2024年在中国三大终端六大市场销售额超过15亿元。在具有明显临床优效性的GLP-1RA方面,1类新药HEC88473正在开展II期临床,为国内研发进展最快的GLP-1/FGF21双重激动剂;生物类似药利拉鲁肽注射液步入III期临床,该产品2024年在中国三大终端六大市场的销售额超过17亿元。在具有较大临床需求的NASH药物方面,东阳光药布局了FXR激动剂、GLP-1/FGF21双靶激动剂、THR-β激动剂、ACC抑制剂等主流靶点。其中,HEC96719片(FXR激动剂)、HEC88473注射液(GLP-1/FGF21双靶激动剂)已处于II期临床,HEC169584胶囊(THR-β激动剂)、HEC138671片已获批临床。资料来源:米内网数据库、CDE官网等注:米内网《中国三大终端六大市场药品竞争格局》,统计范围是:城市公立医院和县级公立医院、城市社区中心和乡镇卫生院、城市实体药店和网上药店,不含民营医院、私人诊所、村卫生室,不含县乡村药店;上述销售额以产品在终端的平均零售价计算。数据统计截至4月30日,如有疏漏,欢迎指正!免责声明:本文仅作医药信息传播分享,并不构成投资或决策建议。本文为原创稿件,转载文章或引用数据请注明来源和作者,否则将追究侵权责任。投稿及报料请发邮件到872470254@qq.com稿件要求详询米内微信首页菜单栏商务及内容合作可联系QQ:412539092【分享、点赞、在看】点一点不失联哦
▎来源:药明康德内容团队
3月21日,中国国家药监局(NMPA)官网最新公示,联邦制药以注册分类3.3类申报的利拉鲁肽生物类似药上市申请已获得批准。根据联邦生物早先新闻稿,该产品为一款胰高血糖素样肽-1(GLP-1)类似物,本次获批的适应症为糖尿病。
截图来源:NMPA官网
GLP-1是肠道细胞分泌的一种多肽类激素,它通过与GLP-1受体相结合,刺激胰岛素的分泌,并且抑制胰高血糖素的分泌,从而促进葡萄糖的代谢。同时,它还能够能起到延缓胃排空和抑制食欲的效果。天然的GLP-1在人们用餐之后,受到葡萄糖的刺激而分泌。而在2型糖尿病患者中,GLP-1的分泌水平显著下降,这也是导致患者血糖控制不佳的重要原因之一。
利拉鲁肽原研药由诺和诺德(Novo
Nordisk)开发,已在全球领域获批用于成人2型糖尿病患者控制血糖、超重和肥胖症成人长期体重管理等。
中国药物临床试验登记与信息公示平台显示,联邦制药已经就利拉鲁肽注射液完成两项临床试验,其中一项是在中国健康受试者中开展的药动学和安全性比对试验;另一项是治疗2型糖尿病的有效性和安全性的3期试验。
2型糖尿病(T2DM)是一种慢性代谢疾病,该疾病的根本原因是胰岛β细胞衰竭或丧失,无法分泌足够的胰岛素以保持机体的血糖稳态。GLP-1类似物能够以葡萄糖依赖方式作用于胰岛β细胞,增加胰岛素分泌;刺激β细胞的增殖和分化;抑制食欲及摄食,延缓胃内容物排空;能够有效地控制糖尿病患者血糖,并减少低血糖风险。
参考资料:
[1]2025年03月21日药品批准证明文件送达信息 . From https://www.nmpa.gov.cn/zwfw/sdxx/sdxxyp/yppjfb/20250321152845131.html
[2] 联邦制药利拉鲁肽注射液上市申请获受理. Retrieved Aug 23 , 2023.
From https://mp.weixin.qq.com/s/GsvDyZyy3rFkaDy9Pvhj5Q
药渡媒体商务合作
媒体公关 | 新闻&会议发稿
张经理:18600036371(微信同号)
点击下方“药渡“,关注更多精彩内容
免责声明
“药渡”公众号所转载该篇文章来源于其他公众号平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
微信公众号的推送规则又双叒叕改啦,如果您不点个“在看”或者没设为"星标",我们可能就消散在茫茫文海之中~点这里,千万不要错过药渡的最新消息哦!👇👇👇
100 项与 利拉鲁肽生物类似药(珠海联邦制药) 相关的药物交易