预约演示
更新于:2025-05-07
DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHY 46
46型发育性癫痫性脑病
更新于:2025-05-07
基本信息
别名 DEE46、DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHY 46、Developmental and Epileptic Encephalopathy 46 + [3] |
简介 An autosomal dominant condition caused by mutation(s) in the GRIN2D gene, encoding glutamate receptor ionotropic, NMDA 2D. It is characterized by developmental delay and intractable seizures. |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

NCT03166215
A Phase 1b/2a Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Dose-Escalation Study With an Open-Label Part to Examine the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of TAK-935 as an Adjunctive Therapy in Subjects With Developmental and/or Epileptic Encephalopathies
100 项与 46型发育性癫痫性脑病 相关的临床结果
登录后查看更多信息
100 项与 46型发育性癫痫性脑病 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-03-01Epilepsia
Incidence of childhood and youth epilepsy: A population‐based prospective cohort study utilizing current International League Against Epilepsy classifications for seizures, syndromes, and etiologies
Article
作者: Brandlistuen, Ragnhild E. ; Chin, Richard F. ; Aaberg, Kari M. ; Vikin, Truls ; Lossius, Morten I.
2023-09-01Stem Cell Research
Generation of a human iPSC line (ZJSHDPi001-A) from peripheral blood mononuclear cells of a patient with Developmental epileptic encephalopathy-47 carrying FGF12 gene mutation (c.334G > A)
Article
作者: Guo, Yufan ; Miao, Pu ; Wang, Ye ; Feng, Jianhua ; Lou, Yuting ; Shi, Xinglei ; Gao, Liuyan ; Su, Guofa
2022-06-01Epilepsia1区 · 医学
International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions
1区 · 医学
Article
作者: Gwer, Sam ; Cross, Helen J. ; Yozawitz, Elissa ; Hirsch, Edouard ; Zuberi, Sameer M. ; Pressler, Ronit ; Wiebe, Samuel ; Tinuper, Paolo ; Specchio, Nicola ; Auvin, Stéphane ; Wilmshurst, Jo M. ; Riney, Kate ; Perucca, Emilio ; Wirrell, Elaine C. ; Moshé, Solomon L. ; Guerreiro, Marilisa ; Scheffer, Ingrid E. ; Nabbout, Rima ; Samia, Pauline
分析
对领域进行一次全面的分析。
登录
或
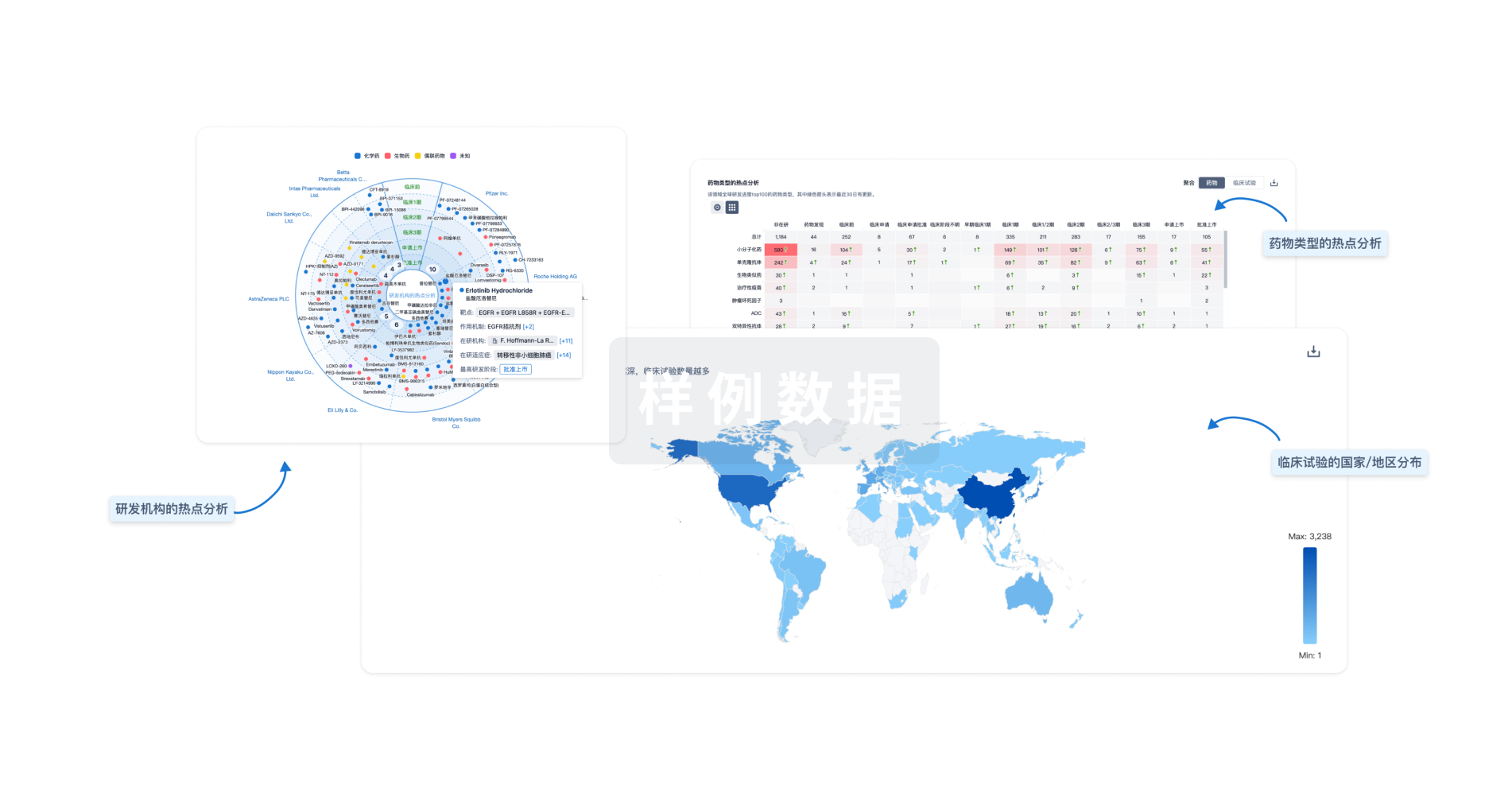
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用