预约演示
更新于:2025-05-07
Unspecified blepharoconjunctivitis
未特指的睑结膜炎
更新于:2025-05-07
基本信息
别名 BLEPHARO CONJUNCTIVITIS、BLEPHAROCONJUNCTIVITIS、Blepharo conjunctivitis + [12] |
简介 Inflammation of both the eyelids and the conjunctiva. |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

CTRI/2022/08/044716
A Multi Centre, Randomized, Double Masked, Placebo-Controlled, Parallel-Group, Phase III Comparison Study to Assess Efficacy and Safety of Bibrocathol 2% Eye Ointment (Posiformin 2%) in the treatment of Chronic Blepharoconjuctivitis. - NA
NCT03975374
Efficacy and Safety of Tobradex Eye Drop Suspension vs Tobramycin/Dexamethasone Ophthalmic Suspension
ISRCTN14084351
A multi-centre, randomized, double-masked, placebo-controlled, parallel-group, phase III study to assess efficacy and safety of Bibrocathol 2% eye ointment (Bibrocathol-POS 2%) in the treatment of chronic blepharoconjunctivitis

100 项与 未特指的睑结膜炎 相关的临床结果
登录后查看更多信息
100 项与 未特指的睑结膜炎 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-05-01DNA Repair
Identification of a novel pathogenic XPC:c.2420 + 1 G>C variant in a patient with xeroderma pigmentosum
Article
作者: Faatih, Mukhlissul ; Ulhaq, Zulvikar Syambani ; Ratnangganajati, Estu
2025-03-01Ophthalmic Plastic & Reconstructive Surgery
An Atypical Mpox Presentation With a Large, Chronic Right Upper Eyelid Ulcerative Mass
Article
作者: Moreau, Annie ; Do, Thai ; Khattab, Mostafa ; Pierre, Dakota St. ; Steinberger, Elise E.
2024-05-16Cureus
Bilateral Ophthalmomyiasis Externa of Lid by Musca domestica: A Rare Presentation
Article
作者: Sune, Mona P ; Sune, Manjiri P ; Mahajan, Shital M ; Sune, Pradeep
2024-07-14
上市批准生物类似药
分析
对领域进行一次全面的分析。
登录
或
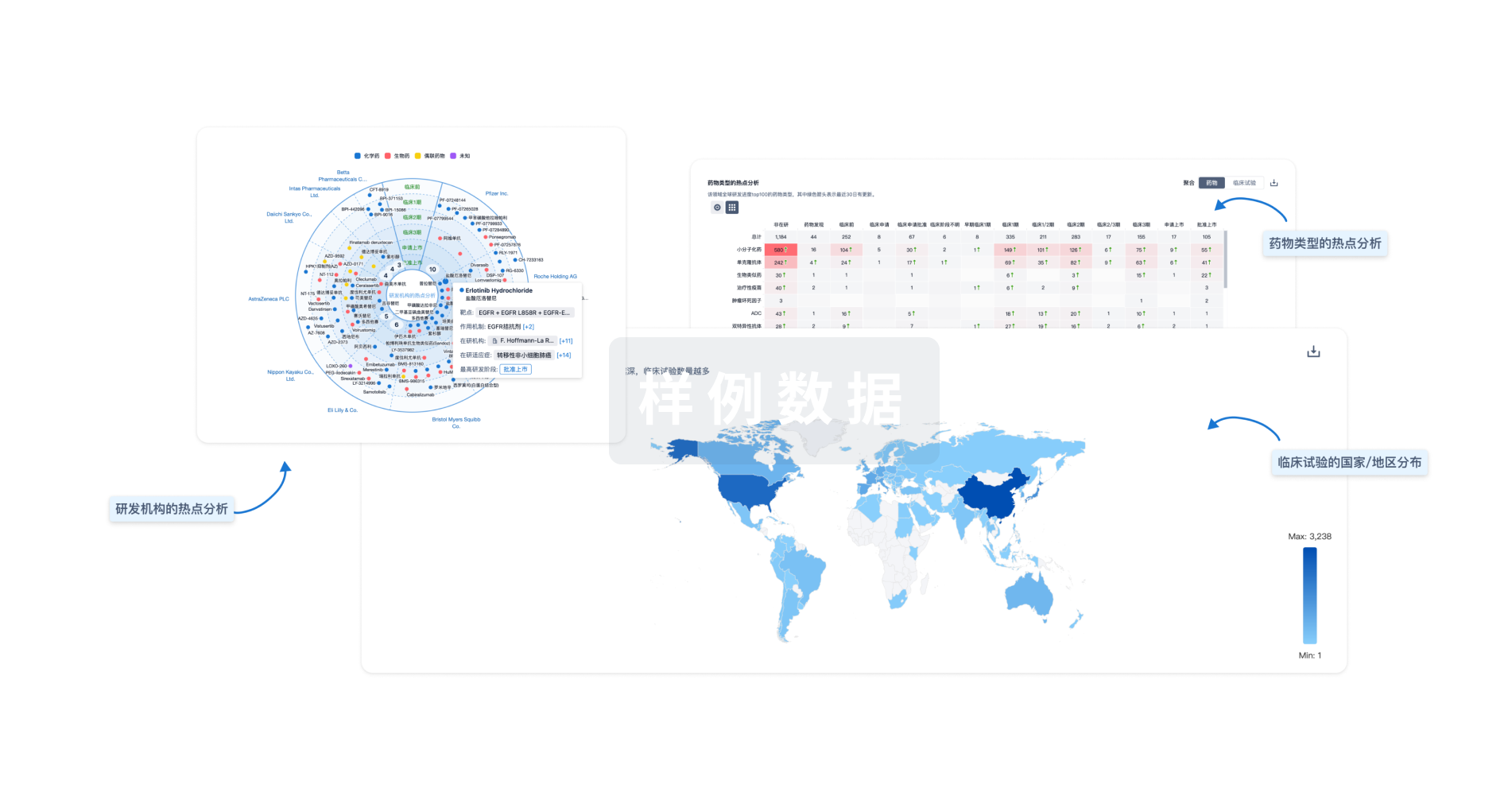
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用