更新于:2024-11-01
CYP8B1
更新于:2024-11-01
基本信息
别名 7-alpha-hydroxy-4-cholesten-3-one 12-alpha-hydroxylase、7-alpha-hydroxycholest-4-en-3-one 12-alpha-hydroxylase、CYP12 + [7] |
简介 A cytochrome P450 monooxygenase involved in primary bile acid biosynthesis. Catalyzes the 12alpha-hydroxylation of 7alpha-hydroxy-4-cholesten-3-one, an intermediate metabolite in cholic acid biosynthesis (PubMed:10051404). Controls biliary balance of cholic acid and chenodeoxycholic acid, ultimately regulating the intestinal absorption of dietary lipids (By similarity). Mechanistically, uses molecular oxygen inserting one oxygen atom into a substrate, and reducing the second into a water molecule, with two electrons provided by NADPH via cytochrome P450 reductase (CPR; NADPH--hemoprotein reductase) (By similarity). |
关联
100 项与 CYP8B1 相关的临床结果
登录后查看更多信息
100 项与 CYP8B1 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2024-11-01Drug Metabolism and Disposition
CYP8B1 Catalyzes 12alpha-Hydroxylation of C27Bile Acid: In Vitro Conversion of Dihydroxycoprostanic Acid into Trihydroxycoprostanic Acid
Article
作者: Yu, Tingting ; Zhang, Xinjie ; Chen, Qi ; Wang, Yixuan ; Wang, Yutong ; Wu, QingLiang ; Hu, YiTing ; Zeng, Wushuang ; Lan, Ke ; Gui, Lanlan
2024-10-01Cell Metabolism
Hepatic FXR-FGF4 is required for bile acid homeostasis via an FGFR4-LRH-1 signal node under cholestatic stress
Article
作者: Yang, Miaomiao ; Huang, Zhifeng ; Xu, Da ; Hou, Yushu ; Tang, Ruqi ; Luo, Yongde ; Liu, Yi ; Hu, Yue ; Ma, Jianjia ; Shi, Keqing ; Xie, Cen ; Tang, Yuli ; Chen, Jie ; Rao, Zhiheng ; Zheng, Minghua ; Huang, Zhuobing ; Luo, Jianya ; Song, Lintao ; Lu, Mingqin ; Dai, Xijia ; Cai, Chao ; Li, Xiaokun ; Chen, Chuchu ; Ma, Xiong
2024-10-01Journal of Biological Chemistry
Development of a high throughput cytochrome P450 ligand-binding assay
Article
作者: Scott, Emily E. ; Frydendall, Elyse ; Scott, Emily E
2022-09-09
·生物谷
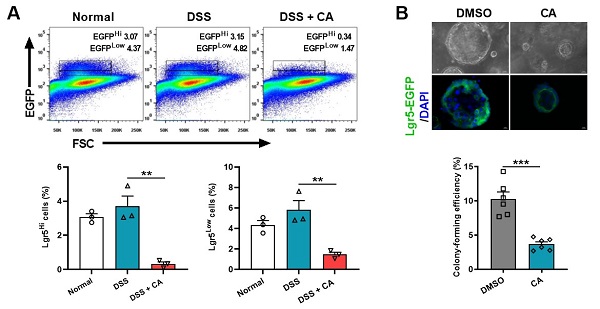
2022-09-05
分析
对领域进行一次全面的分析。
登录
或
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用