预约演示
更新于:2025-06-13
Beijing Stonewise Technology Co., Ltd.
更新于:2025-06-13
概览
标签
肿瘤
小分子化药
疾病领域得分
一眼洞穿机构专注的疾病领域

技术平台
公司药物应用最多的技术
靶点
公司最常开发的靶点
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
100 项与 北京望石智慧科技有限公司 相关的临床结果
登录后查看更多信息
登录后查看更多信息
2025-12-31JOURNAL OF ENZYME INHIBITION AND MEDICINAL CHEMISTRY
Identification of broad-spectrum M
pro
inhibitors: a focus on high-risk coronaviruses and conserved interactions
Article
作者: Liu, Man ; Zhang, Wei ; Liu, Shun ; Peng, Wei ; Fang, Yuying ; Wang, Shunjing ; Sun, Zeyun ; Zhao, Li ; Zhang, Hongbo ; Tang, Zhenhao ; Huang, Xupeng ; Rao, Zixuan ; Zhong, Yihang ; Yu, Linyi ; Yan, Mengrong
The COVID-19 pandemic underscores the urgent need to develop broad-spectrum antivirals against coronaviruses (CoVs) to prepare for future outbreaks. In this study, we presented a systematic approach to developing broad-spectrum Mpro inhibitors, with a focus on high-risk CoVs. We optimised S-217622 as a lead compound, with the goal of enhancing conserved interactions within the S1, S2, and S3/S4 pockets of Mpro, leading to significantly improved inhibitory potency against representative CoVs. Compound 25 exhibited submicromolar activity across all ten CoVs, with IC50 values below 0.1 μM for six of them. The X-ray co-crystal structure of SARS-CoV-2 Mpro in complex with compound 25 revealed the structural basis of conserved interactions contributing to its broad-spectrum activity. This study demonstrates the feasibility of reinforcing conserved interactions to develop Mpro inhibitors with broad-spectrum activity and provides valuable strategies for combating future pandemics caused by unknown CoVs.
2025-05-01EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY
Expanding the utilization of binding pockets proves to be effective for noncovalent small molecule inhibitors against SARS-CoV-2 Mpro
Article
作者: Zhang, Hongbo ; Dai, Jun ; Huang, Bo ; Wei, Dandan ; Liu, Man ; Liu, Lijie ; Yan, Mengrong ; Rao, Zihe ; Yang, Qi ; Huang, Xupeng ; Zhong, Yihang ; Chen, Zhao ; Shi, Yongxia ; Zhao, Jincun ; Tang, Zhenhao ; Wang, Yiliang ; Zhang, Wei ; Peng, Wei ; Sun, Zeyun ; Zhu, Airu ; Wang, Dong ; Zhao, Yao ; Zhao, Li ; Tang, Jielin ; Sun, Jing ; Yang, Haitao ; Chen, Xinwen
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and continues to pose serious threats to global public health. The main protease (Mpro) of SARS-CoV-2 is crucial for viral replication and its conservation, making it an attractive drug target. Here, we employed a structure-based drug design strategy to develop and optimize novel inhibitors targeting SARS-CoV-2 Mpro. By fully exploring occupation of the S1, S2, and S3/S4 binding pockets, we identified eight promising inhibitors with half-maximal inhibitory concentration (IC50) values below 20 nM. The cocrystal structure of Mpro with compound 10 highlighted the crucial roles of the interactions within the S3/S4 pockets in inhibitor potency enhancement. These findings demonstrated that expanding the utilization of these binding pockets was an effective strategy for developing noncovalent small molecule inhibitors that target SARS-CoV-2 Mpro. Compound 4 demonstrated outstanding in vitro antiviral activity against wild-type SARS-CoV-2 with an EC50 of 9.4 nM. Moreover, oral treatment with compounds 1 and 9 exhibited excellent antiviral potency and substantially ameliorated virus-induced tissue damage in the lungs of Omicron BA.5-infected K18-human ACE2 (K18-hACE2) transgenic mice, indicating that these novel noncovalent inhibitors could be potential oral agents for the treatment of COVID-19.
2025-01-01COMPUTERS IN BIOLOGY AND MEDICINE
Deep multiple instance learning on heterogeneous graph for drug–disease association prediction
Article
作者: Kang, Hongyu ; Jiang, Rui ; Li, Jiao ; Zhang, Bowen ; Zheng, Si ; Gu, Yaowen
Drug repositioning offers promising prospects for accelerating drug discovery by identifying potential drug-disease associations (DDAs) for existing drugs and diseases. Previous methods have generated meta-path-augmented node or graph embeddings for DDA prediction in drug-disease heterogeneous networks. However, these approaches rarely develop end-to-end frameworks for path instance-level representation learning as well as the further feature selection and aggregation. By leveraging the abundant topological information in path instances, more fine-grained and interpretable predictions can be achieved. To this end, we introduce deep multiple instance learning into drug repositioning by proposing a novel method called MilGNet. MilGNet employs a heterogeneous graph neural network (HGNN)-based encoder to learn drug and disease node embeddings. Treating each drug-disease pair as a bag, we designed a special quadruplet meta-path form and implemented a pseudo meta-path generator in MilGNet to obtain multiple meta-path instances based on network topology. Additionally, a bidirectional instance encoder enhances the representation of meta-path instances. Finally, MilGNet utilizes a multi-scale interpretable predictor to aggregate bag embeddings with an attention mechanism, providing predictions at both the bag and instance levels for accurate and explainable predictions. Comprehensive experiments on five benchmarks demonstrate that MilGNet significantly outperforms ten advanced methods. Notably, three case studies on one drug (Methotrexate) and two diseases (Renal Failure and Mismatch Repair Cancer Syndrome) highlight MilGNet's potential for discovering new indications, therapies, and generating rational meta-path instances to investigate possible treatment mechanisms. The source code is available at https://github.com/gu-yaowen/MilGNet.
100 项与 北京望石智慧科技有限公司 相关的药物交易
登录后查看更多信息
100 项与 北京望石智慧科技有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2025年06月17日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
临床申请批准
1
登录后查看更多信息
当前项目
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
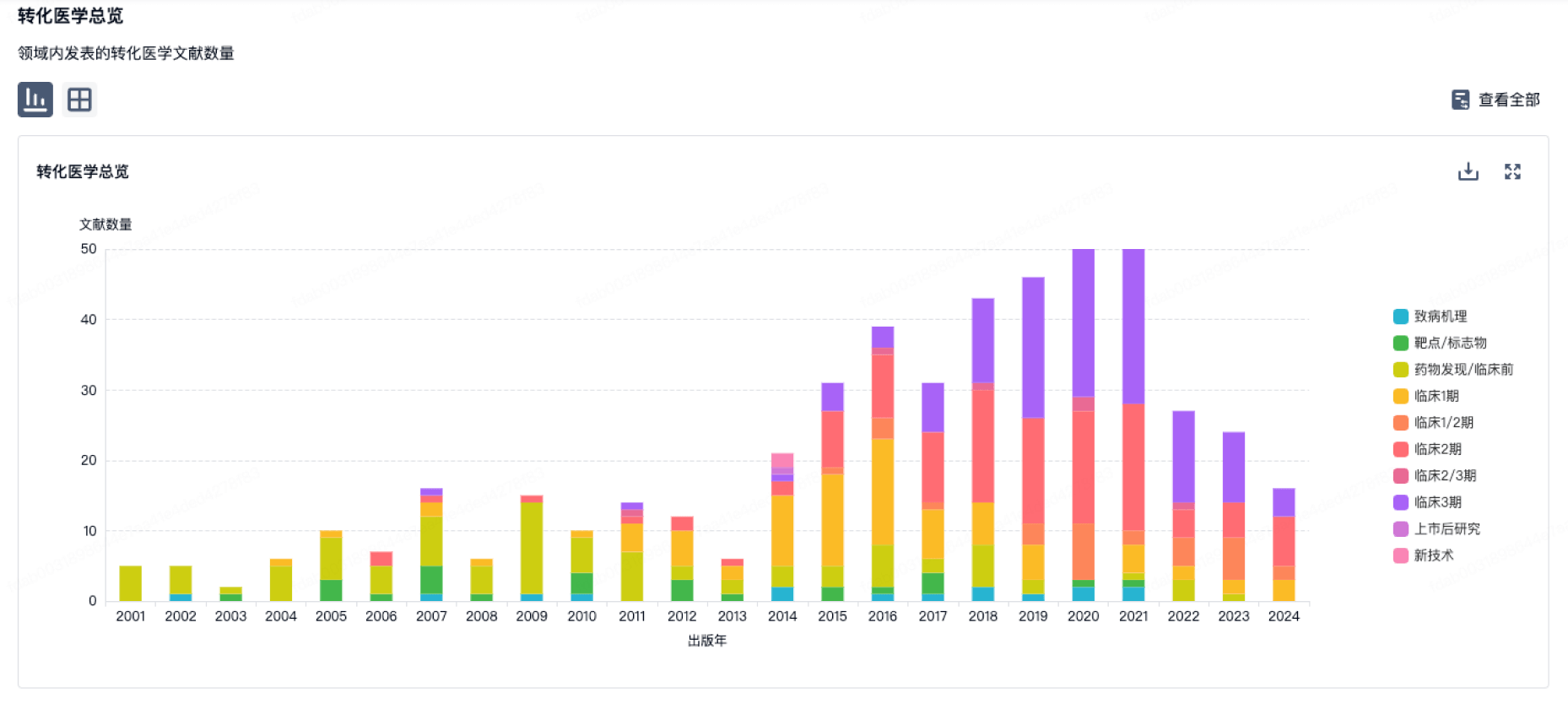
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用