更新于:2024-05-01
New Bio Co., Ltd.
私营公司|
South Korea
私营公司|
South Korea
更新于:2024-05-01
概览
关联
100 项与 New Bio Co., Ltd. 相关的临床结果
登录后查看更多信息
0 项与 New Bio Co., Ltd. 相关的专利(医药)
登录后查看更多信息
2
项与 New Bio Co., Ltd. 相关的新闻(医药)2022-11-15
ADC已成为一类很有前途的抗肿瘤药物,目前已有十几种ADC药物获得批准,用于治疗多种类型的癌症患者。传统的 ADC 利用抗体赖氨酸的氨基或打开链间二硫键获得的半胱氨酸的巯基进行偶联,然而其均一性差,稳定性低,影响了药效及治疗窗口。为了改进这些问题,研究人员开发了各种特定部位的定点偶联方法。这些方法将细胞毒素或化疗药物偶联到抗体分子中特定的位置,例如半胱氨酸、谷氨酰胺、非天然氨基酸、短肽标签和多糖,从而制备均一度高,稳定性好,具有更好的活性和药代学特性的ADC。本文就四大类经典的定点偶联技术在 ADC 中的应用做一个简单介绍。通过特定氨基酸实现位点特异性偶联几种天然或工程改造的氨基酸,包括半胱氨酸和谷氨酰胺,被选作位点特异性的偶联位点。THIOMAB技术是第一个对天然氨基酸进行修饰,将未配对的半胱氨酸进行位点特异性的方法。该方法将半胱氨酸残基插入抗体重链(HC)或轻链(LC)的不同位置进行偶联(图1)。由于工程半胱氨酸在表达过程中易被谷胱甘肽或其他覆盖,抗体需要部分还原以去除帽子。然后,使用硫醇-马来酰亚胺化学方法,将未封端的半胱氨酸与含有硫醇连接子进行反应。研究表明通过半胱氨酸残基(HC-A114C)的药物连接子偶联产生的ADC显示出近乎均一的偶联物,并且改善了治疗指数。例如,抗MUC16 TDC(THIOMAB-drug conjugate)在小鼠卵巢癌移植瘤模型中的体内效果比用传统半胱氨酸法制备的ADC在同等药物的剂量下药效提高了一倍。大鼠和食蟹猴对TDC的耐受剂量也高于常规ADC。图1. THIOMAB技术产生的TDC后续研究者开发了将药物连接子与抗体特定设计的半胱氨酸进行定点偶联的ADC。目前,多个位点特异性的ADC已进入临床,如SGN-CD19B、CD123A和CD352A(均为HC-S239C突变);RG7861/DSTA4637S(LC-V205C突变);IMGN632(S442C突变);BAT8003(A114C突变);以及基于双半胱氨酸突变的ADC,如PF-06804103(LC-K183C HC-K290C)。还开发了两种其他的技术,即半胱氨酸插入,如MEDI2228(HC-i239C);以及HC-末端多肽融合,如ALT-P7(C-末端ACGHAACGHA融合)。然而,半胱氨酸工程抗体的使用并不能保证临床成功,一些已经放弃开发,例如,BAT8003和上面提到的Seagen的 三个ADC。除了通过未配对的半胱氨酸偶联外,还发展了硫醇桥联方法。每个双功能药物连接子可以捕获两个游离的半胱氨酸硫醇基团,从而在所有八个链间二硫键完全还原后导致DAR4 ADC,异质性较低(图2)。例如,New Bio 的 NBT828。图2. 双马来酰亚胺试剂交联半胱氨酸谷氨酰胺通过位点特异性偶联也被报道。该方法不使用还原和氧化试剂,而是利用谷氨酰胺转氨酶(MTGase)将含胺的药物连接子或反应性spacer转移到HC-Q295脱糖抗体中(图3)。利用该技术的Innate ADC(IPH43)药物目前处于临床前阶段。图3. 利用谷氨酰胺转氨酶(mTG)将含胺的药物连接子或反应性spacer转移到脱糖抗体中通过非天然氨基酸实现位点特异性偶联药物连接子与抗体中非天然氨基酸的偶联是另一种新型定点偶联产生匀质性ADC的方法。目前引入的非天然氨基酸通常为乙酰苯丙氨酸(pAF)、叠氮基甲基-L-苯基丙氨酸(pAMF)和叠氮赖氨酸(图4)。引入非天然氨基酸的抗体与药物连接子可定点、定量偶联,获得 DAR 均一、药效高、稳定性好、安全性高的 ADC,但也存在抗体表达困难,易产生免疫原性的弊端。图4. 通过非天然氨基酸实现位点特异性ADCAmbrx 公司的 ARX788 是首个利用非天然氨基酸开发的抗体偶联药物,目前处于临床研究阶段。ARX788选择的非天然氨基酸是乙酰苯丙氨酸(pAF),pAF上的酮基可与有效载荷AS269上的羟胺基团形成肟键,发生特异性位点的偶联,产生均质的ADC。Sutro Biophma开发了一个无细胞蛋白表达系统,通过特定位置掺入p-叠氮甲基-L-苯丙氨酸(pAMF),适合于随后药物连接子点击化学的偶联以此制备ADC。由于无细胞蛋白表达的效率,进行了位点扫描以确定最佳偶联位置,目前已被用于各种ADC项目。例如, STRO-001(DAR2 ADC,HC-F404的偶联)、STRO-002(DAR4 ADC, HC-Y180 和 HC-F404 中的偶联)。通过糖工程实现位点特异性偶联通过将药物连接子与位于CH2结构域的N297糖链偶联,糖链介导的偶联提供了一种独特的位点特异性偶联方法。由于糖链的非还原末端存在几种不同的单糖,人们开发了不同的方法将药物连接子连接到这些糖上,包括岩藻糖、半乳糖、N-乙酰半乳糖胺(GalNAc)、N-乙酰氨基葡萄糖(GlcNAc)和唾液酸(SA)。Okeley等人报道了6-硫代果糖(一种岩藻糖类似物)可以通过代谢偶联到抗CD30或抗CD70抗体中。然后将抗体中的硫代果糖与含有MMAE药物连接子的马来酰亚胺偶联,DAR为1.3。硫代果糖产生的ADC保持了良好的血浆稳定性,并显示出较强的抗肿瘤活性。半乳糖或半乳糖类似物也被通过半乳糖基转移酶引入。菌株促进的环炔与叠氮化物的环加成反应已被用于GlycoConnect,这是Synaffix开发的一项技术,其核心是将叠氮糖以酶促方式引入到天然抗体的多糖上。抗体首先在UDP 6-叠氮基GalNAc存在下经受内切糖苷酶和糖基转移酶(GalNAc-T) 两种酶的作用,以便将天然糖转化为均质的、截断的和叠氮标记的三糖(图5)。接下来,对叠氮标记的抗体进行药物连接子偶联,以产生均一的ADC。GlycoConnectt技术目前被用于三个临床ADC药物:ADCT-601(ADC Therapeutics)、XMT-1592(Mersana Therapeutics)和MRG004A(Miracogen)。图5. UDP 6-叠氮基GalNAc存在下用内切糖苷和GalNAc-T进行多糖建模,通过连接有效载荷的无金属点击偶联获得均一的ADC此外,也开发了利用唾液酸(SA)进行了位点特异性偶联的方法。SA首先被转移到抗体上,然后用高碘酸氧化以连接到含氨氧基的药物连接子(图6)。用此方法制备的抗HER2 ADC具有较强的体外和体内抗肿瘤活性。另一种类似的方法是通过无铜点击化学将C9-叠氮化SA转移到到抗体上,然后与含有细胞毒素的DBCO偶联。图6. 利用唾液酸(SA)进行的位点特异性偶联反应糖工程方法的独特之处在于,药物连接子与糖链偶联,而不需要设计氨基酸序列,并且它们连接在远离氨基酸残基的地方。然而,该方法需要糖工程所需的特殊试剂和酶。通过短肽标签实现位点特异性偶联目前开发了一些通过将细胞毒素与含有四到六个氨基酸残基的特定短肽标签偶联的位点特异性偶联方法。Strop等人的研究将谷氨酰胺标签(LLQG)设计成抗体分子,标签中的谷氨酰胺可以被MTG识别以转移含胺药物。该研究证明,药物连接子MMAD可以通过酶有效地转移到谷氨酰胺标签上,包括C端重链上的LLQGA或C端轻链上的GGLLQGA。该ADC具有均一性的 DAR 2,展现了强效的抗肿瘤活性。另一项研究通过使用转肽酶介导的转肽反应,产生位点特异性偶联的ADC。转肽酶的主要功能是帮助蛋白质附着到细菌细胞壁上,并组装菌毛。转肽酶作用于含有 C 端细胞壁分选信号的分泌蛋白,该信号含有一个五残基识别基序,例如 LPXTG,可以与特定的寡甘氨酸受体底物发生酰胺化反应 ,从而用受体的寡甘氨酸片段取代LPXTG的末端甘氨酸。这个方法已经用于产生ADC(图7)。例如,NBE Therapeutics公司利用这一方法,通过将LPETG标签融合到重链C-末端,然后与五甘氨酸修饰的细胞毒有效载荷PNU-159,682连接,开发了已经进入临床的ADCs 药物NBE-002。同样,GeneQuantum开发了基于重链 C 端 LPGTG 的GQ-1001药物。图7. 转肽酶介导的Gly5-Linker有效载荷与C端LPXTG标记抗体的偶联这类方法依赖于将独特的短肽标签引入抗体中进行酶修饰。虽然这类方法很简单,但这些短肽标签的潜在免疫原性目前尚不清楚,需要后续更多的临床验证。小编小结通过将细胞毒素或化疗药物与工程特定的氨基酸、非天然氨基酸、短肽标签和N297多糖偶联,研究人员开发了下一代位点特异性偶联技术。这些方法产生的ADC具有高度均一性,保证了生产过程批次间的重现性,与传统的偶联物相比具有更高的治疗指数。毫无疑问,这些新型技术未来将会为ADC药物更多的突破和成功发挥重要作用。参考文献1.Site-Specific Antibody Conjugation for ADC and Beyond.2.Chemical Linkers in Antibody–Drug Conjugates (ADCs).3.Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index.4.Site-Specific Antibody−Drug Conjugation through Glycoengineering.5.Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index.6.Enzymatic Antibody Modification by Bacterial Transglutaminase.7.Transglutaminase-Based Chemo-Enzymatic Conjugation Approach Yields Homogeneous Antibody−Drug Conjugates8.凯莱英药闻公众号
抗体药物偶联物临床结果AACR会议
2022-09-12
Linker-drug (LD) 与抗体的偶联是产生ADC和确定ADC关键质量属性的关键步骤,其可以从根本上改变ADC的药代动力学和治疗指数。目前超过95%的ADC基于两种偶联技术:半胱氨酸烷基化或赖氨酸酰化。前十个获批的ADC中有7个基于半胱氨酸偶联(Adcetris、Polivy、Padcev、Enhertu、Trodelvy、Blenrep和ZynLonta);3个基于赖氨酸偶联制备(Kadcyla、Mylotarg和Besponsa)。除了传统的这两种方法外,目前新的技术正在迅速涌现,不仅涉及半胱氨酸(烷基化和交联化)和赖氨酸(酰化或烷基化)偶联的创新方法以产生更加均质的ADC并提高治疗指数,而且还扩展到新的偶联技术,例如基于羰基、酰胺、叠氮基的偶联技术。本文将分两个部分对ADC的这些偶联技术做一个介绍。巯基(-SH)巯基(-SH)是ADC领域中最普遍的反应之一,主要基于三个元素。(A) 巯基是最亲核的氨基酸侧链官能团(比胺更亲核);(B)抗体中适合结合的链间半胱氨酸的数量相对较少(对于IgG1和IgG4为8个),并且对抗体的稳定性不重要,这使得相对可控制的DAR成为可能;(C)链间半胱氨酸,存在于铰链区,连接重链(HC)和轻链(LC),在连接后可以‘隐藏’疏水有效载荷。目前基本上硫醇偶联 ADC 均基于马来酰亚胺化学(图 1)。已经开发了多种马来酰亚胺类试剂用于烷基化,烷基化发生在天然链间半胱氨酸或工程半胱氨酸上。图1.基于马来酰亚胺化学偶联ADC硫醇的基团马来酰亚胺的烷基化反应马来酰亚胺是顺丁烯二酸酐与胺衍生物反应的结构元素,很容易通过亲核Michael加成硫醇基团而发生烷基化反应,形成稳定的硫醚键(图2)。马来酰亚胺在pH为6.5-7.5范围与硫醇反应,其反应速度是胺反应的1000倍,从而可以用最小的试剂量实现完全转化。马来酰亚胺烷基化的主要缺点是Michael加成的可逆性,其速率高度依赖于它所连接的特定半胱氨酸残基的pKa。这种逆Michael反应可能导致连接子药物(LD)在循环中过早释放,并可能导致不良事件,这也一直是开发更稳定的马来酰亚胺变种的重要驱动因素。图2.各种马来酰亚胺试剂与半胱氨酸烷基化单抗中的半胱氨酸都参与了二硫键的形成,因此需要在与LD结合之前进行还原步骤以释放硫醇基团。通过优化还原剂(通常为TCEP或DTT)的量,以释放特定的自由硫醇基团(对于IgG1和IgG4,最多释放8个)。通过与半胱氨酸偶联制备的上市的ADC有Adcetris、Polivy、Padcev、Enhertu、Blenrep、Trodelvy、Zynlonta、RC48等。由于缺乏位点特异性,这些ADC通常以随机混合物的形式产生,平均DAR在2.3-4之间。然而,有两个例外,Enhertu和Trodelvy是通过所有链间半胱氨酸的接近定量水平结合产生的,DAR分别为7.7和7.6,因此具有位点特异性。尽管在绝大多数使用马来酰亚胺-己基连接子。然而有一些电子不足的氨基乙基或乙二醇间隔基的ADC可能抑制逆迈克尔反应,例如基于β-氨基乙基的AbGn-107、Zynlonta、ADCT-502、IMGN632、JBH492、MEDI2228和TR1801-ADC;基于乙二醇的SYD985、MORAb-202和ZW49。一些苯基取代的马来酰亚胺,据报道也更稳定,目前还处于临床前开发中。后续研究者开发了将LD与抗体特定设计的半胱氨酸进行定点结合的ADC,相比随机ADC混合物,其可能会提供更高的治疗指数。目前,多个位点特异性的ADC已进入临床,如SGN-CD19B、CD123A和CD352A(均为HC-S239C突变);RG7861/DSTA4637S(LC-V205C突变);IMGN632(S442C突变);BAT8003(A114C突变);以及基于双半胱氨酸突变的ADC,如PF-06804103(LC-K183C HC-K290C)。还开发了两种其他的技术,即半胱氨酸插入,如MEDI2228(HC-i239C);以及HC-末端多肽融合,如ALT-P7(C-末端ACGHAACGHA融合)。然而,半胱氨酸工程抗体的使用并不能保证临床成功,一些已经放弃开发,例如,BAT8003和上面提到的Seagen的 三个ADC。通过对硫醚-琥珀酰亚胺加合物进行水解以提供顺丁烯二酸衍生物,可以抑制不希望发生的逆Michael反应。这种水解可以通过在pH为9的条件下对ADC进行长时间的处理来实现;然而,这可能导致蛋白质降解事件,例如脱酰胺或焦谷氨酸的形成。Seagen通过使用DPR技术,在烷基链的a位上带有氨甲基,解决了此问题 (图3)。在加入硫醇后,氨基诱导琥珀酰亚胺结合物的自发自水解,从而阻止了逆Michael反应。引入该技术的ADC产品有SGN-CD48A(现已停产)、 SGN-CD228A。图3. 半胱氨酸与基于二氨基丙酸(DPR)氨甲基的马来酰亚胺试剂进行烷基化反应,然后自水解得顺丁烯二酸偶联物其他半胱氨酸烷基化试剂各种替代马来酰亚胺烷基化的技术近年来也被开发出来。例如,a-卤代乙酰胺试剂的高反应性是众所周知的,其中 2-碘乙酰胺是游离半胱氨酸封端的无可争议的可选择试剂。更重要的是,其形成的硫醚键的是完全不可逆的,因此在稳定性方面优于硫醇-马来酰亚胺醚。另外Alley的研究使用a-溴乙酰胺丙基连接子为硫醚ADC提供了极好的血浆稳定性,在小鼠体内注射2周内没有可测到的系统性药物释放。由于甲磺酰基苯并噻唑(MSBT)是一种选择性的硫醇封闭剂,Barbaset等人开发了一系列芳香族甲磺酸试剂,用于硫醇蛋白质的烷基化反应。类似地,甲磺酸基嘧啶与硫醇通过亲核芳香取代机理进行反应(图4),这一概念应用于SKB264。Glythera(现为 Iksuda)的研究人员报告了通过将游离半胱氨酸硫醇基团与 4-乙烯基吡啶反应获得的高度稳定的硫醚 ADC,这种技术称为 PermaLinks(图4)。所得 ADC 具有被证明具有良好的稳定性。柏林大学开发的亚膦酸膦技术,现在已被Tubulis商业化使用,其与传统的马来酰亚胺相比,具有缺电子三键的芳基膦酰亚胺化合物显示出极好的半胱氨酸选择反应活性,同时所得到的(Z)-乙烯基硫醚具有极好的稳定性(图4)。图4. 半胱氨酸烷基化的替代技术双马来酰亚胺或双马来酰胺酸烷基化/交联化马来酰亚胺烷基化的能力也被应用于各种交联双马来酰亚胺试剂(图5)。每个试剂将捕获两个游离的半胱氨酸硫醇基团,从而在所有八个链间二硫键完全还原后导致DAR4 ADC。例如,New Bio 的 NBT828 中使用了双马来酰亚胺交联剂。图5. 双马来酰亚胺试剂交联半胱氨酸来自阿斯利康的研究人员报道了用双马来酰亚胺修饰的有效载荷(PBD)重桥半胱氨酸工程抗体,以获得DAR1 ADCs(图6)。图6. 双马来酰亚胺修饰的有效载荷重桥半胱氨酸工程抗体β-sulfone的烷基化/交联反应游离硫醇的高亲核性和简便易行的Michael加成也被用于不同类型的交联剂中,最初被Del Rosario和Wilbur报道,并由Poly-Therics (现为Abzena;THIObridge技术)为ADC开发。重桥可以通过使用双硫醇反应基团重新连接形成的硫醇来增加还原抗体的稳定性,从而更好地保持最终连接物中的构象完整性。用由双-a,a-甲苯磺酰基甲基酮前体原位形成的α-甲苯磺酰基甲基-α,β-不饱和酮试剂处理还原抗体会导致一系列反应,例如,Michael加成,β-消除 甲苯基亚磺酸,并再次Michael加成以提供稳定的交联产物(图7)。THIObridget 技术目前用于 OBI999 ADC(phase 1 阶段)。图7. 半胱氨酸与甲苯磺酸丙烯酸酯试剂的交联(THIObridge技术)双溴甲基试剂的烷基化/交联反应第三种DAR4 ADCs的交联方法是由Concortis (现为Sorrento)以C-lock技术开发的。C-lock技术结合了硫醇的亲核性和溴化苄基亲核SN2取代。因此,通过还原链间二硫化物,然后与双溴甲基芳基试剂反应,例如双溴甲基喹恶啉,发生不可逆交联制得硫醚(图8)。一款使用C-lock技术的ADC(CD38-ADC) 已经进入临床。图8. 半胱氨酸与双溴甲基喹恶啉的双烷基化交联(C-lock)。二溴并氮杂二酮的烷基化/交联化受二溴马来酰亚胺基团对还原二硫键重新桥联的启发, Chudasamaa等人开发了一种基于氮杂二酮(PD)基团的核心结构的的重桥技术。与马来酰亚胺衍生物不同,氮杂二酮环更稳定,不会通过水解开环,从而得到更均匀的共轭化合物。研究者开发了一溴PD(MBPD)和二溴PD (DBPD)的衍生物,并证明了这两种衍生物都能够在pH 8.0的条件下与硫醇基团发生特异性反应。使用DBPD硫醇重桥化学技术已被用于创建具有高度受控的有效载荷修饰的ADC(图9)。图9. 抗体硫醇基团与二溴并氮杂二酮衍生物的交联二硫键的形成虽然不直接连接到抗体半胱氨酸上,但ADC早期开发中就应用了二硫键作为连接子的一部分,例如MLN2704、CMD-401、BIWI-1和IMGN242。在细胞毒有效载荷和抗体之间含有二硫键的连接子设计可以推动可逆连接,这种连接可以通过肿瘤细胞内的谷胱甘肽还原来裂解以释放毒素。然而,由于溶解的半胱氨酸的存在,循环中二硫键的过早断裂经常发生,这导致进入肿瘤细胞之前释放有效载荷。在二硫化物附近的碳原子上添加保护基团制造空间位阻来增加循环的稳定性,但在许多情况下,它们也会降低肿瘤细胞内毒素的释放速度。此外,即使受阻二硫化物的稳定性增加,ADC也不如药物结合在抗体序列中最佳位置的工程半胱氨酸残基上有效。Pillowet等人是第一个将小分子药物直接连接到抗体不同位置的工程半胱氨酸上(图10)。通过使用受阻或不受阻二硫键的ADC进行比较,确定LC-K149C位置上的工程半胱氨酸是结合含有二硫键的药物最稳定的结合部位。使用LC-K149C位点创建的ADC表现出极好的循环稳定性,同时允许肿瘤细胞内谷胱甘肽的有效切割。图10. 通过工程半胱氨酸产生二硫键以制备抗体结合物氨基 (–NH2)氨基天然存在于赖氨酸侧链中,是与单抗偶联最方便的官能团,其不需要预先的化学修饰步骤。虽然由于其高的pKa(~10.5),大部分蛋白质在中性pH下被质子化,但蛋白质中仍有一小部分氨基将以中性、未质子化的形式存在。氨基在亲核性方面仅次于硫醇基团,但明显领先于其他的氨基酸的侧链(如精氨酸、丝氨酸、色氨酸、蛋氨酸)。而且,赖氨酸的氨基不像半胱氨酸硫醇那样需要还原释放,并且大量可用,例如,抗体可能包含多达80个暴露在溶剂中的赖氨酸残基。胺类化合物修饰的初级偶联反应通过两种主要的途径:酰化或烷基化。一般来说,这些反应是快速和选择性的,产生稳定的键(酰胺键或仲胺键),并导致高产率。然而,由于赖氨酸的大量存在,有限数量的选择性酰化是非常具有挑战性的,因此赖氨酸偶联的ADC通常显示出广泛的DAR分布,结合发生在大量的位点上(图11)。图11. 通过酰化(A)或还原烷基化(B)与天然赖氨酸发生随机偶联随机酰化羟基丁二酰亚胺(NHS)酯是最常用的用于氨基酰化反应的羧酸的活性衍生物。NHS含酯试剂很容易在碳二亚胺存在下由羧酸盐和NHS反应生成,随着NHS leaving基团的释放,与胺类亲核试剂反应生成稳定的酰胺键(图12)。这种酯与其他亲核试剂,如醇或水的竞争反应慢了几个数量级,而与硫醇或组氨酸氮基的反应产生不稳定的硫酯或咪唑基酯,很容易水解或仍与邻近的胺交换形成酰胺键。除了中性的NHS酯外,还可以制备具有更好水溶性的磺化衍生物(sulfo-NHS)。例如,在制造Kadcylas中使用的SMCC试剂。图12. 氨基与NHS酯的酰化反应通过赖氨酸酰化制备的代表性ADC有 Mylotarg、Besponsa、IMGN853和SAR408701。法国斯特拉斯堡大学的Wagneret等人报告了一种通过4-叠氮苯甲酰氟(ABF)与赖氨酸氨基反应的新方法。该试剂与抗体上的伯胺快速反应形成酰化产物,比使用基于NHS酯的试剂效率更高(图13)。其末端叠氮基可以用于偶联毒素、荧光染料和寡核苷酸。图13. 4-叠氮苯甲酰氟与赖氨酸侧链反应生成叠氮化抗体位点特异性酰化虽然赖氨酸侧链的直接酰化是抗体偶联的最直接和最容易的方法,但广泛的DAR分布的是其一个显著的缺点。因此,开发了位点特异性酰化技术。例如,Concortis Biosystems 公司(现为Sorrento Therapeutics)报道了一种名为K-lock的一步赖氨酸结合技术,该技术可以通过将细胞毒性有效载荷连接到预先选择的抗体位点来产生位点特定的ADC。尽管该技术的细节尚未披露,但已经被用于各种项目,例如,与 Lova Therapeutics合作的靶向5T4的 ADC药物(ZV05)。Bernardes等人也报道了特定部位的赖氨酸酰化反应。使用从计算机辅助预测的具有最低pKA的抗体赖氨酸的试剂,在略碱性pH的酰化反应将在动力学上有利的。研究发现磺酰基丙烯酸酯仅使用单摩尔当量(37℃,pH 8.0)就能快速进行反应,使得曲妥珠单抗轻链Lys207的定量和不可逆修饰,并完全保留天然二级结构和功能(图14)。图14. Lys207与磺酸甲酯的反应,然后氨基官能化的有效载荷的aza-Michael加成Ajinomoto的研究人员报道了一种两阶段、定点赖氨酸偶联Ajicap技术。这项技术的核心是精心设计的来自protein A的高亲和力环肽,其与抗体的Fc部分具有高亲和力。研究发现,如果环肽带有反应性的酯部分,则抗体-多肽复合体附近的Lys248的酰化反应具有极高的选择性。随后还原二硫键部分,同时从抗体中去除亲和肽,并释放出一个自由的硫醇基团,用于与有效载荷进行反应(图15)。图15. 基于抗体Fc与高亲和肽结合的Ajicap技术官能团相互转化(NH2 to SH)BMS的BMS-986183和BMS-936561(MDX-1203)优雅地结合了氨基和硫醇的化学成分。用 Traut’s试剂(一种五元亚氨基硫酯)处理抗体会导致赖氨酸上amidine(脒)基团的形成、环打开和游离硫醇的暴露(图16)。除去多余试剂后,用马来酰亚胺官能化的接头反应很容易得到所需的ADC。显然,用这种方法制备的ADC,虽然有趣,但可能同时具有赖氨酸偶联的广泛分布和硫醇基团潜在的逆Michael反应的缺点。图16. 抗体与Traut试剂反应,引入游离的硫醇基团,通过基于马来酰亚胺的连接子进行偶联HTI-1066/SHR-A1403(恒瑞)采用了一种将氨基转化为硫醇的替代方法。首先使用S-(3-氧丙基)硫代乙酸酯处理c-Met抗体,使其在天然赖氨酸上安装了游离的硫醇基团。然后通过马来酰亚胺共轭引入有效载荷(图17)。图17. S-(3-氧丙基)硫代乙酸酯与抗体的赖氨酸烷基化反应引入游离的硫醇基团,通过马来酰亚胺共轭引入有效载荷官能团相互转化(NH2 to马来酰亚胺)最常见和成功应用的官能团相互转化方法之一是双功能活性酯/马来酰亚胺试剂SMCC或其磺化变体(磺化SMCC)来处理天然赖氨酸。然后通过Michael加成来引入硫醇功能化的连接子-有效载荷(图18)。或者,如果用硫醇部分适当官能化,则可以引入有效载荷自身,例如,DM1就是这种情况,已应用于多种ADC,最著名的是Kadcyla(T-DM1)、DEBIO-1562和Avid100。图18. 首先用SMCC试剂处理天然抗体,然后添加硫醇功能化的有效载荷,制备ADC参考文献1.Chemical Linkers in Antibody–Drug Conjugates (ADCs).2.Characterization of disulfide bond rebridged Fab–drug conjugates prepared using a dual maleimide pyrrolobenzodiazepine cytotoxic payload.3.Site-specific conjugation of native antibodies using engineered microbial transglutaminases.4.Structure and dynamics of a site-specific labeled Fc fragment with altered effector functions.
抗体药物偶联物抗体合作寡核苷酸小分子药物
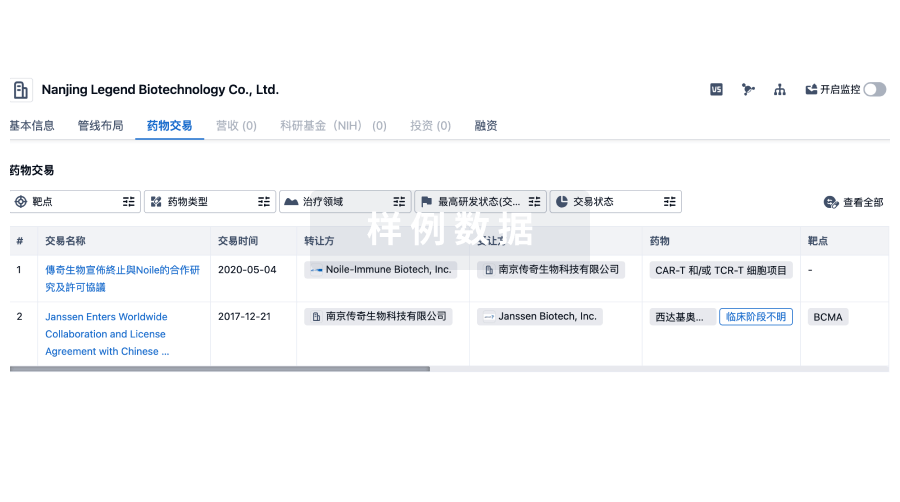
100 项与 New Bio Co., Ltd. 相关的药物交易
登录后查看更多信息
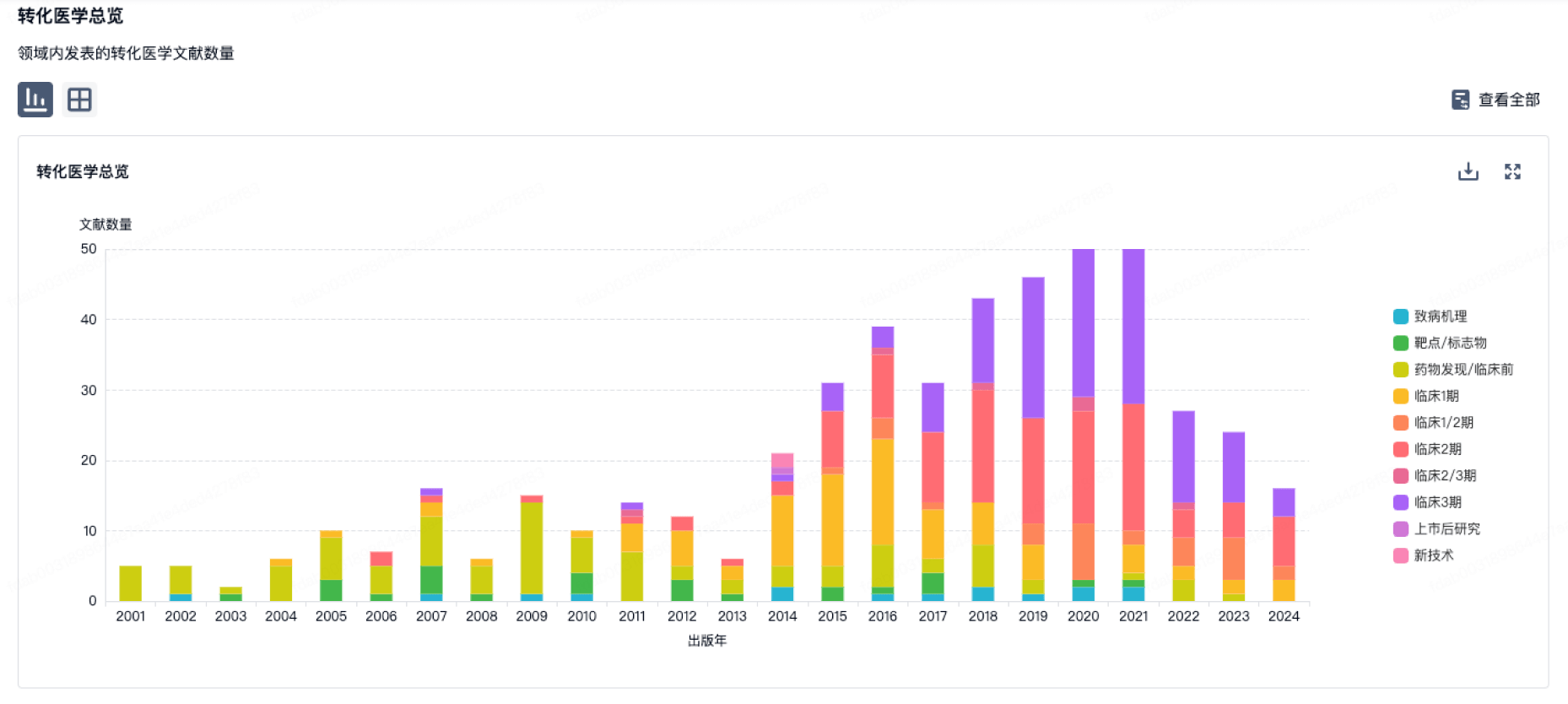
100 项与 New Bio Co., Ltd. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年07月05日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用