更新于:2024-06-24
Blas Lorente Gonzalez SL
更新于:2024-06-24
概览
关联
100 项与 Blas Lorente Gonzalez SL 相关的临床结果
登录后查看更多信息
0 项与 Blas Lorente Gonzalez SL 相关的专利(医药)
登录后查看更多信息
2
项与 Blas Lorente Gonzalez SL 相关的新闻(医药)2022-07-13
抗体药物概论及我国市场发展现状抗体的定义为由抗原(一般为外来蛋白质)刺激后由免疫细胞产生的能与抗原发生特异性反应的免疫性球蛋白,而只针对某一特定的抗原决定簇起作用的抗体称为单克隆抗体(简称单抗)。抗体与靶抗原结合具有高特异性、有效性和安全性。临床用于恶性肿瘤、自身免疫病等各种重大疾病,如果按照其特异性结合抗原或其表位的数量进行分类,抗体药物可分为单克隆抗体、双特异性抗体,甚至有一些三抗或四抗正在研发中。而我国的药典和指导原则对单抗和双抗有明确的定义。人用重组单克隆抗体制品,系指采用各种单克隆抗体筛选技术、重组DNA技术及细胞培养技术制备的单克隆抗体治疗药物,包括完整免疫球蛋白、具有特异性靶点的免疫球蛋白片段、基于抗体结构的融合蛋白、抗体偶联药物等。其作用机制是通过与相应抗原的特异性结合,从而直接发挥中和或阻断作用,或者间接通过Fc效应子发挥包括抗体依赖和补体依赖细胞毒作用等生物学功能。图:单克隆抗体示意图双特异性抗体(bispecificantibody,BsAb,以下称为“双抗”)是通过细胞融合或重组DNA技术制备的人工抗体,可以同时特异性结合两种抗原或同一抗原的两个不同表位。BsAb能够分别识别和结合两种不同的抗原或抗原表位,所以它可以把免疫细胞等效应细胞或细胞因子连接到肿瘤细胞上,进而增强对靶细胞的杀伤作用;可以结合同一肿瘤细胞上的不同抗原表位以增强其结合特异性,同时减少脱靶毒性带来的不良反应;或者结合同一免疫细胞上不同的免疫检查点,同时阻断/激活下游免疫信号通路,激活或抑制免疫细胞。常见的一些双抗技术平台结构示意图(Antibodies 2020, 9, 0065)根据目前FDA已经获批的抗体药物分析,截至2021年6月30日,获批的总数按照靶点和抗体类型进行拆分,FDA共批准122种单抗药物上市,适应症多为抗肿瘤。随着技术的进步以及各类型平台的不断成熟,双抗、ADC以及多抗等多种药物的市场不断扩大。例如双抗在2016-2020以120.4%的复合年增长率快速发展,多抗预期将在2020-2025年迎来增长高峰期,但单抗仍占较大比重。在我国尽管抗体药物产业起步较晚,但是随着近年来高表达细胞株构建,细胞大规模培养工艺,表征分析与质控技术等行业技术壁垒的突破,越来越多的国产抗体药物开始注册临床试验,并成功上市。从适应症和靶点角度来看,我国现有的单抗涵盖的适应症较少,且还存在大量未满足的临床需求。随着研发和审批程序加快,抗体类药物有望覆盖包括心血管、眼科及神经系统在内的更多治疗领域。从抗体药物技术研发角度来看,为了降低抗体药物的免疫原性,抗体开发平台从最开始的鼠源一步步发展,人鼠嵌合,人源化,到全人源抗体。此外,随着DNA重组技术的快速发展,在抗体筛选,scFc工程等方面取得了重大突破,使得例如半衰期延长形式的抗体,增强生物活性的抗体药物等功能多样化的抗体药物得到研发,抗体的开发也逐步走向了,人源化,功能化,小型化,专业化。从国内抗体药物竞争角度来看,我国单抗药物靶点同质化较高,大部分市场份额被跨国公司占据,拥有已被证实的临床价值的创新产品管线的公司应从竞争环境中脱颖而出。抗体药物的法规及过程CMC考量重点相较于基因治疗产品,抗体药物相关法规和政策都推出较早,例如,2001年3月FDA颁布了题为《Monoclonal Antibodies Used as Reagents in Drug Manufacturing》的GMP指南,该指南侧重于在NDAs,ANDAs,INDs,BLAs申报过程中的化学,生产,控制(CMC)问题。指南指出,对于单抗试剂,主要重点是评估以下重点因素:1.生物安全性,特别是评估单抗试剂与来自细胞底物或细胞系来源的外源和/或工艺相关杂质的污染。2.单克隆抗体试剂在原料药生产过程中的性能特征(如对目标分子的活性和特异性)。3.最终原料药和/或药品中可能存在单抗试剂的残留量。CDE团队此前在中国临床药理学杂志中发布的文章也列举了8个需要注意的关键点:临床试验用药物的制备;细胞库;生产用原材料;生产工艺和过程控制;病毒灭活/去除验证;质量研究;稳定性研究;容器密闭系统。此外,由于越来越多的双特异性抗体簇拥上市,同时也对双抗药物的CMC问题,临床和非临床开发问题提出了严格要求。顺应这一现象而生的是CDE和FDA关于双抗药物的指南。2021年5月21日,FDA发布了题为《Bispecific Antibody Development Programs Guidance for Industry》的行业指南。除了一般考虑因素外,该指南还对双特异性抗体开发项目的具体监管、质量、非临床和临床考虑因素提供了建议。主要讨论了双抗在CMC中的独特方面以及临床和非临床开发程序。其中在CMC质量考量方面,指南指出,由于双抗的存在形式多种多样,抗原特异性、亲和力、有效性、与其相关的杂质(如同源二聚体或同源错配物种)以及半衰期、稳定性等都是影响其质量所要考量因素。从IND到BLA的药学评价创新型抗体药物研发进程具有“创试性”(临床试验存在较大的失败风险)、“阶段性”(药学研究可分阶段展开)和“渐进性”(临床期间通常发生工艺变更)的特征,研发过程中常常伴随着失败风险。①创新型单抗药物例如,创新型单抗药物的药学研究随临床试验进程“分阶段”、“渐进式”的开展,药学评价同样遵从药物研发规律。在申请临床阶段药学评价应重点关注受试者的“安全性”, 而注册生产阶段则应全面保证药品的工艺稳健性与质量可控性,符合监管方对于药物内涵的定义。如:“安全、有效和质量可控”或“安全(safety)、纯度(purity)和效力(potency)”。单抗药物在注册生产阶段其药学研究内容应基本完成,与申报临床阶段相比,药学评价在细胞基质、工艺表征和验证、质量研究和控制等方面均提出符合商业化生产阶段特点的更高要求,如:工程细胞系在申报临床阶段可以仅构建主细胞库,特殊情况下甚至是未经单克隆化的细胞池(cell pool),但在申报上市阶段应规范建立三级细胞库, 生产细胞一般应符合“单克隆化”要求。②工艺开发工艺开发方面,申报临床阶段可直接采用平台化的中试工艺,临床期间进行产品特异化的工艺开发。注册上市阶段生产工艺应完成商业化生产工艺的性能确认与工艺验证。③质量研究质量研究方面,申报临床阶段对于产品相关变体(分子大小、电荷异构、翻译后修饰等)可采用报告含量的方式进行初步控制,注册生产阶段则需明确产品相关的杂质或物质,并将限度要求纳入质量控制;同时,应建立反映临床作用机制的生物测活方法并纳入放行标准。稳定性试验考察结果,临床试验阶段只要能够满足临床试验用药的质量控制即可,上市阶段则需要提供全面的稳定性考察数据支持上市产品的有效期和储存、运输条件;病毒去除/灭活工艺,在临床试验申请时重在有效工艺步骤的初步验证,注册生产上市时应完成全面的验证。此外,对于抗体药物生产过程中所接触到高风险的一次性耗材与惯用包材,在产品注册生产时也应完成产品特异性相容研究。④改良型抗体而对于一些在单抗药物基础上进行改良的改良型抗体,其设计依据在于通过分子改构,克服原型蛋白临床局限性。因此,其药学评价应重点关注其“分子改构”的目的、方法与效果。上游构建方面应结合立题依据,在详述分子改构的原因、突变位点的选择依据。质量研究中应结合原型抗体进行比较研究,从结构确证、免疫学活性、生物活性等方面验证分子改构符合预期。如果缺乏与原型抗体(或已上市抗体)的质量比较数据,或证实上述改构不具有功能学意义,是不支持其作为“创新药”生物制品进入临床试验的。⑤双特异性抗体与传统抗体相比,双特异性抗体改变了既往单抗“双价、单靶标”的结合模式。因此,此类创新抗体的质量研究应重点对多个活性区域的结构与功能进行研究,如:肽图中应说明结合不同靶标的轻、重链氨基酸序列及complementarity-determining region特征肽段;功能学研究应说明不同靶标的结合能力和生物活性。此外,还应关注双特异性抗体区别于天然抗体的独特性质。如:BiTE型抗体由于不含恒定区、体内半衰期较短和T细胞激活功能的临床使用剂量小(μg级),临床使用过程中需使用特殊溶解方式和给药装置。图:BiTE型抗体结构示意⑥复方抗体复方抗体是多个单克隆抗体的混合物,可识别病毒颗粒的不同表位,提高临床上的保护效力。由于复方抗体的有效成分为多种单抗,其药学资料应包括提供不同抗体的原材料、生产工艺、质量研究,以及复方制剂的混合方式。质量研究中应结合不同抗体结合表位、生物活性,说明复方组成的立题依据。对于制剂灌装前进行混合的复方抗体,应关注半成品混合工艺的控制;对于单独包装、临用混合的复方抗体,应关注混合配伍后的临床使用稳定性。如图为几款埃博拉抗体鸡尾酒 图片来自:doi.org/10.1038/s41573-022-00495-3小编总结总的来说,尽管抗体类药物经过多年发展,其监管体系等方面已经比较成熟,但越来越多的新型抗体技术的出现使得抗体申报又面临许多新的挑战,例如对于新冠中和抗体的加速审批考量,多抗药物一些重要临床参数的加入,申报阶段临床数据可比性,此外互联网时代eCTD使得申报更需要专业性,可能需要配套软件等等。以信达的信迪利单抗中美申报遭遇的挑战来看,使得临床多元化这一要求可能阻遏抗体药物的成功申请。在美国申报之外,欧盟、日本等地也是药品出海的重要市场。近年来,欧盟质量受权人(Qualified Person)质量审计对抗体药物生产提出高要求。如果某一药企需要把抗体产品的临床样品运到欧洲,需要被欧盟认可的Qualified Person来放行,否则临床产品进不了欧洲市场,这可能对药企造成许多不必要的麻烦。值得一提的是,2021年11月,金斯瑞蓬勃生物南京抗体蛋白药生产基地顺利通过欧盟QP质量审计,标志着金斯瑞蓬勃生物南京抗体蛋白药生产基地已经符合欧盟GMP标准,有能力为国际客户提供高质量的临床样品生产和CMC服务。这是金斯瑞蓬勃生物南京抗体蛋白药生产基地首次欧盟QP质量审计,是对其质量体系符合国内和国际法规要求的持续性验证。截至目前,金斯瑞蓬勃生物南京生产基地已通过来自中国、美国、亚太和欧洲的客户和第三方的多次审计。该欧盟QP审计流程依照Eudralex Vol4(欧盟GMP)法规以及ICH等指导原则,审核范围涵盖质量管理体系、生产管理体系、物料管理、设施设备管理、质量控制、验证和计算机化系统等管理体系。QP审计报告称,金斯瑞蓬勃生物南京生产基地具备高标准的质量管理体系、高度符合标准的设施设备、经验丰富的员工。审计官给予金斯瑞蓬勃生物高度评价,其生产场地完全符合欧盟的监管标准。金斯瑞蓬勃生物GMP生产车间遵循国际领先的设计理念,是真正意义上的“零交叉,单向流”生产线,满足FDA、EMA和NMPA监管要求。车间整体采用了严格的物理隔离措施,可同时进行多种产品的生产。目前,金斯瑞蓬勃生物已经助力抗体药领域客户获得全球临床批件15个。此外,金斯瑞蓬勃生物根据不同目标市场客户和监管要求(包括NMPA,FDA,EMA,ICH和WHO)设计并建立了综合和高效的质量合规体系,以满足不同市场,不同国家的需求。参照PDA TR56,开发并建立特有的与产品生命周期各阶段相适应的质量合规体系(PAC),同时也开发并建立了金斯瑞蓬勃生物特有的CQV客户质量需求管理平台,专注于质量、产品生命各阶段合规和交付价值达成甚至超越客户期待,以合规和高效理念,平衡成本和效率,实现快速申报。图:金斯瑞蓬勃生物CQV管理体系为推动中国创新药高质量发展,助力中国创新药走向更广阔的国际化舞台造福全球患者,2022第三届QbD生物药质量科学大会(点击了解详情)将于8月12-13日在京隆重召开。会议召开前一天,由金斯瑞蓬勃生物主办、佰傲谷承办的“金斯瑞蓬勃生物创新药质量研究案例分析闭门会”将于2022年8月11日在北京朝阳悠唐皇冠假日酒店召开,QbD会前质量研究经典案例剖析,质量高管齐聚一堂,抽丝剥茧,层层深入,既有经验总结,又有新思路启发,我们诚挚的邀请并满心期待着您的到来。参考来源:宋丽娜, 崔靖, 何伍. 创新单抗药物申报临床阶段药学研究关注点[J]. 中国临床药理学杂志, 2021, 37(10):4.刘伯宁, 徐刚领, 罗建辉. 关于我国单抗药物上市阶段药学评价的思考[J]. 药学学报, 2019, 54(11):9.刘伯宁, 罗建辉. 关于创新型抗体药物药学评价的思考[J]. 药学学报, 2017, 52(12):9.金斯瑞蓬勃官网FDA官网EMA官网点击“阅读原文”,进入官网立即报名
创新药基因疗法抗体加速审批抗体药物偶联物
2021-02-28
·药事纵横
摘要本文作者:魏利军,药事纵横创始人 雅培总部位于芝加哥,通过100多年的厚积薄发,该公司已经在2010年成为了全球顶级医药巨头,然而就在最鼎盛的时期,该公司被拆分成了新雅培和艾伯维。从表面上看,雅培似乎在“背道而驰”,分出了最赚钱的创新药业务就等于整个公司被掏空,但事实上,分家以后两个公司都得到了非常好的发展,如今艾伯维已经成为全球前十大制药巨头,而新雅培却成为最具潜力的公司,市盈率高达200多……雅培/艾伯维的发家史极具传奇色彩,研究他人的历史可以预示我们的未来,本文是笔者在研究雅培/艾伯维发家史过程中的笔记,内容含有大量的个人观点,请读者理性阅读,如有错误,欢迎指正。本文不允许其他任何形式的媒体私自转载,如需转载须获得作者本人授权。
❈❈
早期的磅礴发展
Abbott laboratories(雅培公司)的历史可以追溯到1888年,创始人是一位名为Wallace Calvin Abbott的芝加哥医生。在十九世纪末期,人类的现代制药工业还没有成型,虽然当时人们已经可以使用到多个单一成分的药品,但基本都提取自天然药物,如吗啡、奎宁、士的宁和可待因等生物碱,因为还没有完善的现代化制剂工艺,市面上流通的药品主要是汤剂。汤剂因为运输不方便,药物的稳定性也受到影响,作为医生的Abbott,对这种现状非常苦恼。后来比利时医生将这些生物碱药物做成了片剂,虽然质量同样不是很可靠,但Abbott博士看到了希望,于是他打算自己做药,创办了雅培药厂。
那个时候美国还没有《食品、药品和化妆品法》,因此药品上市前也不需要审批,产品基本都是药店或小作坊生产出来的,这对于那些有志者而言可以“说干就干”,而且立杆就能见影,产品马上就能上市销售。1888年,年方30岁的Abbott已经是一名执业医师和药店的老板,他开创了一种名为“Dosimetric granules”小丸剂,不但患者服用更加方便,而且剂量更加准确,得到患者的普遍欢迎,在公司成立的当年,营收就达到了2000美元(约合当前的50万美元)。
十九世纪九十年代以后,Abbott开始广泛地向医生同行推广他的产品,因为广泛的临床需求和巨大的产品优势,Abbott的业务得到蓬勃发展。由于在那个时代,药品几乎都是直接面向医生销售,为了加速产品的推广和普及,Abbott还积极地拉医生参股,并在1900年组建了Abbott Alkaloidal Company(雅培生物碱公司),到1905年,这已经是一个年销售额达20万美元的大公司。随着企业规模的不断扩大,产品线也在不断地丰富,该公司在1910年的新产品数量竟高达700余种,分支机构也覆盖到纽约,旧金山,西雅图和多伦多,部分产品更是通过经销商卖到欧洲和印度。
Abbott Alkaloidal Company的产品
二十世纪初期,随着化工工业的高速发展,很多提取药物陆续被化学合成出来,为了体现公司的转型,该公司被改名为Abbott Laboratories,并一直沿用至今。一战爆发以后,雅培开始生产各种战争必须药物,比如抗疟疾药奎宁,伤口护理剂Chlorazene等等。这些产品因为其他公司也在生产,因此并不能体现雅培的重要性,能体现雅培公司重要性的药物是普鲁卡因和巴比妥。由于德国在一战中是美国的敌对国家,开战以后,德国药物的被断供,长期依赖德国药物的美国人面临着严重缺药的境地,尤其是一些具有巨大临床需求的必须药物。在此期间,雅培公司因开发出普鲁卡因(procaine)和巴比妥(barbital)成功替代了德国生产的诺瓦卡因(novocaine)和Veneral,而在美国医学界取得很高的影响力。
1921年,Abbott博士去世,Alfred Stephen Burdick博士被任命为公司的掌门人。在Burdick的主导下,该公司在北卡罗纳州的研发实验室投入运营,陆续开发出多种新药,包括麻醉剂、镇静剂和维生素等。因为积极地研发和推广,雅培公司在二十年代发展非常迅速,1929年,雅培公司登陆芝加哥证券交易所上市,公开发行2万股,每股32美元。1933年,Clough, S. DeWitt成为雅培的新一任CEO,通过Clough积极地改革创新,虽然遭遇经济危机,但该公司依然保持着较高的活力。除此以外,Clough还创办了公司内刊,对员工的舆论进行引导,士气得到很大的提高提振。
自二十年代成功开发出Butyn(丁卡因)之后,雅培公司就与麻醉剂搭上了不解之缘,时至今日,麻醉剂直都是该公司的一大特色。在Butyn之后,雅培又推出了Nembutal(戊巴比妥钠)和Pentothal(硫喷妥钠),其中Nembutal成为该公司史上最长寿的药品,而Pentothal因为太畅销而其开发者在50年之后入选了美国名人堂。二战期间,以麻醉剂为特色的雅培,在战争中发挥了巨大的作用,与此同时,该公司还应美国政府的要求,为战争生产青霉素,成为当时五大青霉素巨头之一。
二战以后,美国政府放开了对青霉素的管制,几乎所有能够量产青霉素的企业都发了一笔不小的横财,1948年,雅培已经是一家拥有1500多名员工的制药公司,生意做到了北美、欧洲和巴基斯坦等国家。然而青霉素没有专利,全球各地的制药企业在短短几年间里,陆续地开始量产,青霉素不再是理想中的肥肉,因此二战以后,雅培公司也积极地从事新型抗生素的开发,其中五十年代最具代表性的产品是Erythrocin(硬脂酸红霉素)和E.E.S.(琥乙红霉素)。
多元化的困局
五十年代,雅培的研究员在一次偶然的工作中发现一种具有甜味的物质——环己基氨基磺酸盐(甜蜜素),最初,这种产品只是向糖尿病患者推广,但六十年代以后,随着美国人变得更加关注健康和饮食,这种物质的商业用途开始逐步扩大。甜蜜素的巨大潜力,为雅培的多元化发展提供了条件和机遇。
甜蜜蜜的化学结构
六十年代以后,雅培制药的特色是大输液和抗生素,因为新药研发领域多年无突破,制药业务的高速发展难以保持,为了降低业绩的压力,于是该公司开始考虑多元化。1964年,雅培收购了营养品巨头M&R Dietetics,获得了奶粉业务,其中包括著名的Similac系列配方奶粉。1967年,Edward J. Ledder被任命为公司CEO,他是一个典型的多元化倡导者。他认为多元化可以让公司减少对药品业务的依赖,也让公司变得更灵活,各部门间更富有弹性,失败业务可以获得繁荣业务的补贴,相互促进以度过共同的危机。在上任后的几年里,雅培推出了一系列的消费品,包括Pream牌无脂奶精、Glad牌橡胶手套、Faultless牌高尔夫球和Sucaryl牌甜味剂等,为了确保消费品部门的有序发展,Ledder还从Revlon(露华浓)公司挖来了销售专家Melvin Birnbaum作为部门负责人。
虽然Ledder理想的“灵活性”逐渐地体现了出来,但新的问题也在逐渐地暴露。因为消费品销售费用太高,盈利能力低下,多元化并没有理想中的完美,甜蜜素是该公司唯一爆发的产品,其在1969年的销售额高达5000万美元,几乎占到该公司消费品业务营收的三分之一。然而人红总是是非多,因为甜蜜素的大范围使用遭到FDA的调查。虽然FDA当时给出的证据受到人们的质疑,但是那些证据依然被定性为甜蜜素致癌。1970年8月,FDA禁止甜蜜素在美国市场销售。
在多元化没有吃到甜头的同时,该公司的医疗用品业务受到激烈的竞争,公司的业绩似乎在泥潭里挣扎,而且让雅培雪上加霜的事情还不止这些,在甜蜜素禁售后的第二年,雅培的大输液因为受到细菌污染而被大量召回,销售额从从1790万美元降至300万美元,而且被判罚款1000万美元。在这一系列的打击让雅培陷入了困境,股价逐渐下滑。
争议CEO下的第二春
因为七十年代初期的发展不顺,雅培的高层受到了普遍的指责,但Robert Schoellhorn却成为一枝独秀。Schoellhorn作为医疗用品的副总裁,他成功预测了未来的发展趋势,在此基础上积极布局,雅培的医疗用品业务在1974-1979年间增长了139%。因为出色的表现,Schoellhorn被提拔为公司的总裁和首席运营官,并在1979年登上了董事长和CEO的宝座。
因为多元化失利,七十年代后期的雅培开始将目光逐渐转移回制药,确立了以药品、营养品和诊断设备为核心的战略方向,主张加强研发、强调效率,严控成本,积极国际化扩张。药品方面,推出了Depakene(丙戊酸)、Tranxene(二甲氯氮卓)和Abbokinase(尿激酶)三种新药。除此以外,雅培还积极与其他公司合作,与当时正在出海的武田成立了合资公司TAP pharma,共同开发和销售药物。营养品方面,该公司利用多年的大输液经验积极地开发维生素疗法,开发了一系列的营养品治疗方案,以加速住院患者的康复,从而降低医疗费用。在二十世纪八十年代,多达65%的住院患存在各种形式的营养不良,为雅培提供了巨大的市场空间。与此同时,婴儿食品和家庭护理品业务也在逐渐增长,在巨大的市场中取得了一席之地。诊断用品方面,雅培也取得相当不错的成绩,该公司开发的电子检测设备大大增强了手动检测的精度和便捷性,广受用户的欢迎。
Schoellhorn成为CEO和董事长之后,进一步加强了研发的投入,并强调对外投资。1982年,雅培推出了7种新药,这7种产品在短短的三年内,销售额就增加到公司营收的五分之一。1987年,Hytrin(特拉唑嗪)获得FDA批准用于妊娠高血压,随后TAP pharma的头孢甲肟和亮丙瑞林也获批上市,制药业务的发展迎来了第二次春天。在Schoellhorn的主导下,雅培的海外业务也得到快速地发展,先后在30多个国家建立起75家子公司或制造工厂。在发展制药的同时,Schoellhorn并没有推倒Ledder的多元化战略,推出了Murine牌眼部护理品和Selsun Blue牌去屑洗发水以扩大消费品管线,并承诺让其它低迷的业务枯木逢春。诊断业务方面,在Schoellhorn的主导下,雅培成为了血液分析仪的主导者,广泛用于血液中各种物质的检测。1985年,雅培推出了针对HIV和HBV的诊断测试设备,到八十年代末,该公司的血液分析仪销售额超过了10亿美元,几乎占到医疗产品营收的一半。
Schoellhorn是雅培历史上非常出色的一位领导人,同时也是一位非常具有争议性的领导人。在他的带领下,雅培七十年代的颓势被彻底改变,销售额在八十年代翻了3倍,而利润翻了2倍,然而遗憾的是Schoellhorn在雅培并没有得到“善终”。因为独特的管理风格,领导层之间的斗争非常激烈,在八十年代的10年中,James L. Vincent、Kirk Raab和Jack W. Schuler等三位总裁离职,而Schoellhorn本人也在1989年12月被董事会驱逐下台,随后,原董事会副主席Duane L. Burnham接管了雅培的帅印。
站在巨人的肩膀上
八十年代的业绩发展为九十年代的腾飞奠定了基础,但在九十年代初期,雅培的制药业务除了丙戊酸钠,依然没有特别像样的产品,1990年的全球药品销售额排名中,雅培排在15名开外,处方药销售额不足20亿美元。尽管如此,该公司发展创新药的决心很明显,从八十年开始,研发投入一直稳步增长,1990年的研发投入达5.7亿美元,与默沙东、BMS等当时的美国一线制药巨头相差无几。
随着研发投入的逐渐增大,雅培在九十年代陆续收获了几个非常有潜力的产品,1991年,新一代红霉素类抗生素Biaxin(克拉霉素)获批上市,1994年吸入麻醉剂七氟烷又获得FDA批准,1997年抗病毒药Norvir(利托那韦)成功推出,尤其是克拉霉素还成为了一个重磅炸弹。除此以外,与武田联合开发的Lupron(亮丙瑞林)和Prevacid(兰索拉唑)持续走俏,每年销售额高达20亿美元。
虽然研发取得了不错的成绩,但是雅培在九十年代的药品销售额并没有实现像八十年代一样的高速增长,一方面是七八十年代推出的产品专利陆续到期,另一方面是研发不再像八十年代那么高产,除此以外,武田在1997年宣布要在美国自建营销管线,不再续签下一个十年的产品代销合同,这将让雅培每年丧失高达20亿美元的销售额。增长乏力,股票低迷,雅培公司面临着被对手吞并的危险,求稳图存的发展道路已经明显走不通了。幸运的是,雅培在九十年代中期,摆脱了保守派领导人的束缚,为了避免被吞并,Burnham主动出击,开启了雅培的并购之路。1996年,雅培以8.67亿美元的价格收购了糖尿病血液检测设备制造商MediSense打响了并购之路上的第一枪,1997年,该公司又花了2亿美元从赛诺菲美国子公司中买下了大输液产品线。
1999年,Burnham退休,诊断部门的高级副总裁Miles D. White接替了他的职位。White是一位能够放开手脚做事情的人,在他的带领下,雅培之前“求稳图存”的发展模式彻底被改变。还在领导权的过渡期,White就以2.34亿美元的价格吃掉了诊断设备制造商Murex Technologies,进一步加强雅培的诊断部门。通过几次医疗和诊断业务的并购,雅培的诊断和医疗用品的销售额在1999年达到52.5亿美元,加之国际业务高速发展,总营收达到132亿美元,相比1990年的62亿美元翻了2倍多。相比之下,虽然收获了几个新产品,但是因为武田不再续签合同的原因,雅培在九十年代的药品销售额并没有大幅上涨,1999年总销售额仅为24亿美元。
为了加强制药业务,1999年6月,雅培宣布以价值73亿美元股票为代价,向载药巨头ALZA发起收购邀约,但遗憾的是这笔交易因雅培的股票暴跌而告吹。收购ALZA失败之后,White很快把目标转向了BASF,最终在2000年以69亿美元的价格拿下BASF的制药部门Knoll,从这笔收购中,雅培获得了Meridia(西布曲明)和Synthroid(左甲状腺素钠)等畅销产品,将雅培药品销售额瞬间提高了20亿美元,与此同时,从这次交易中,雅培还获得一种名为D2E7的在研药物,这就是我们今天所知的神药阿达木单抗。除此之外,White还在跨世纪的一年多时间里完成了多起小型并购,以6亿美元的价格吃掉了手术器械生产商Perclose,以2.2亿美元的价格从GSK买断了5个麻醉药品的销售权。
在BASF的制药业务并入管线之后,雅培很快又将农产品业务出售给住友化学株式会社,成为了纯粹了的医疗保健公司。然而在二十一世纪初期,虽然有来自BASF的三大产品和新艾滋病药物Kaletra(Iopinavir/ritonavir)助力,但雅培的药品销售额一直不足营收的三分之一,2002年仅为42.3亿美元,占总营收的27.6%,然而这一现状很快就因阿达木单抗而发生改变。2002年末,修美乐获得FDA的批准并很快就发展成重磅炸弹,销售额在2005年就达到14亿美元。随着药品销售额渐显起色,雅培在2006年以37亿美元的价格收购了Kos Pharma,换回Niaspan(烟酸控释片)、Flutiform(氟替卡松/福莫特罗)和Advicor(烟酸/洛伐他汀)三大产品,2009年,White又以66亿美元的价格收购了Solvay(苏威),获得Udiliv(熊去氧胆酸)、Duphaston(去氧孕酮)和 Creon(胰酶)等畅销产品,到2010年时,雅培的药品销售额已达200亿美元规模,首次登上了全球十大制药巨头的宝座。除了制药业务,诊断和医疗用品业务也在White的带领下遍地开花,通过持续不断的收购和剥离,雅培的业务活力逐渐爆发了出来,盈利能力也在逐步增强。在二十一世纪的第一个十年间,雅培收购了二十多家相关的公司,两大部门在2010年的销售额近70亿美元,盈利达15亿美元。
雅培分家
White掌权的第一个十年,雅培取得了巨大的成功,销售额翻了2.6倍,净利润翻了4倍,不但让雅培成为了全球前十大制药巨头,还妥善解决了多起“医疗欺诈”和“营销纠纷”案件,获得了美国商界多项至高无上的荣誉。但是在White执掌公司的第一个十年间,雅培的研发投入并没有实质性提升,平均研发投入仅占销售额的9.4%。没有耕耘,自然也就没有收获,在二十一世纪的第一个十年,雅培在自主创新药研发方面,几乎一无所获。并购方面,该公司把目光都放在了产品线的打造上,并没有在研发管线的储备上多下功夫。因为研发不给力,研发管线空虚,药品营收高度依赖阿达木单抗,而该产品将在2016年专利到期,因此,雅培风光的背后潜藏着巨大的隐忧。
雅培要剥离创新药业务的声音,早在2011年就传了出来,这看似奇葩的事情最终还是在2013年成为了现实。通过一年多的股权调整和资产分离,2013年初,创新药部门组建成一家全新的公司Abbvie Inc(艾伯维)上市,原雅培的首席运营官Richard Gonzalez担任CEO和董事长。分家之后,雅培保留了营养品、医疗诊断用品和非专利药品的业务,2013年销售额为218.5亿美元,净利润21.8亿美元,而艾伯维则带走几乎所有的专利药物,包括修美乐、Niaspan、Creon和Tricor等畅销产品,2013年销售额为187.9亿美元,净利润达41.3亿美元。
从数据的表面上看,在分家的过程中,雅培似乎已经被掏空,近十年来的努力都是在给别人做嫁衣,而且把最赚钱的业务剥离出去,无异于自断手足。虽然其它制药巨头也经常剥离业务部门,但基本上剥离的都是盈利能力较低的多元化部门,雅培这种背道而驰到底意欲何为?有分析师曾认为艾伯维的盈利能力虽然比较高,但营收高度依赖阿达木单抗,该产品专利悬崖日益迫近,但该公司研发管线储备非常空虚,阿达木单抗在2016年专利到期之后,该公司的利润将无以为继。除此以外,艾伯维背负着157亿美元的债务,持有资金只有72亿美元,如果按照2012年的现状,该公司要到2015年才能实现财务的平衡。相反,新雅培虽然没有高度赚钱的制药业务,但债务只有75亿美元,持有的资金却达50亿美元,如果稳扎稳打,业绩上升的空间非常大。
说白了,剥离艾伯维,是雅培高层对高投入高风险的制药业务没有足够的信心,想通过拆分将部分债务转嫁到新的公司。但这种观点也有一定的片面性,当时雅培董事长White就进行了解释,拆分是为了让投资者更好地了解两个业务部门,以在差异化的投资中获益。事实上,在拆分之前,雅培的市盈率只有7左右,而拆分之后的一年里,新雅培的市盈率达到21,而艾伯维则达到了17,股东的获益是非常明显的。除此以外,阿达木单抗并没有分析师当时预测的那么“短命”,如今依然为艾伯维带来着丰厚的回报。
事实上长期经营多元化业务的雅培早已经习惯了低风险,长生命周期的多元化生意,虽然利润比较低,但比较把稳。在拆分之后,新雅培卖掉了盈利较低的仿制药业务,收购了CFR Pharma和Veropharm,重新构建了制药部门,2017年药品销售额达43亿美元,不仅如此,根据IMS分析师的预测,雅培在未来5年间,药品销售额的复合增长率高达8.2%,有望在2022年达到65亿美元。诊断和医疗用品方面,雅培卖掉了眼科医疗业务,发动了大量的并购案,收购了10余家公司,在销售额增长的同时,盈利能力稳重有升,2017年的销售额规模达到145亿美元。因为出色的业绩,自2013年以来,雅培的股票高速增长,2017年底的市盈率高达216,远高于艾伯维。
2018年雅培公司的表现评分(Robecosam)
艾伯维因为有巨大的债务,运营上显得有些束手束脚,好在阿达木单抗没有分析师当年预测的那么“短命”,有望在2020年之前持续为该公司带来大把的现金。在拆分之后,艾伯维很快就把研发投入提高的制药巨头的平均水平,与此同时,该公司还积极对外投资,通过合作的形式弥补研发效率的不足。并购方面,Gonzalez也没有松懈,刚完成拆分不久就对夏尔发起了高达540亿美元的收购邀约,但遗憾的是该交易因未获得政府的批准而以失败告终,最终不得不支付16.4亿美元的分手费,这对当时的艾伯维而言,的确是雪上加霜。不过好在失之东隅收之桑榆,艾伯维与Enanta联合研发的丙肝鸡尾酒Viekira在2014年成功上市,投资者对该公司的信心大幅增强,2014年底,艾伯维的市盈率达到53。2015年之后,艾伯维分别以210亿美元和98亿美元为代价,相继收购了Pharmacyclic和Stemcentrx,换回重磅炸弹ibrutinib和在研新药Rovalpituzumab tesirine,除此以外,该公司自研白血病新药Venclexta获得FDA批准,抗肿瘤管线逐渐形成。
经过多年的努力,艾伯维的研发也逐渐有了起色,研发管线中积累了多个比较有潜力的产品,包括妇科新药Elagolix,抗肿瘤新药Veliparib、Depatuxizumab Mafodotin、Rovalpituzumab tesirine和elotuzumab,免疫和炎症新药Upadacitinib和Risankizumab等,在这些产品的助力之下,艾伯维在未来5年里,销售额有望继续高速上涨,复合增长率高达7%,到2022年,该公司的销售额有望达到350亿美元,并登上全球第八大制药巨头的宝座。因为巨大的发展潜力,艾伯维的股票也自拆分以来实现高速上涨,2017年底的市盈率也达到了94。
小结与讨论
企业大了,什么样的人都有,有革新派,自然也会有保守派。从业务的发展上不难看出,二战以来,在雅培的管理层中,明显是保守派占据了上风。保守派治理公司往往“图稳”,为追求低风险高利润,他们会从生产和销售上做文章,积极提高效率,严格控制成本,力争将利润最大化。然而这种利润的提升是局部的,容易受到外部环境的干扰。八十年代以后,虽然CEO Schoellhorn的积极改革颇显成效,但他的管理下,雅培高层动荡不断,3位总裁离职,而他自己也被驱逐出董事会。九十年代以后,雅培虽然逐渐摆脱了保守派的束缚,开始放开手脚进行并购交易,但在高风险的创新药研发上,该公司自始至终都没有放开手脚。
在笔者看来,雅培69亿美元买到阿达木单抗是额外的运气,因为被收购之时,Knoll的营收高达21亿美元,很明显雅培是冲着上市产品去的。然而,雅培制药业务的高速发展却是阿达木单抗带来的,在这运气之外,该公司不愿意去太多的冒险。因此,雅培的拆分可能就是保守派和革新派博弈的一个结果,而拆分本身也是一次巨大的改革。保守派的图稳,低风险、低投入、拼运营成本的营养品、非专利药品和医疗用品再适合他们不过,而相反,高风险,高投入,需要持续不断创新的品牌药业务,用图稳的模式发展肯定是行不通的。除此以外,因为超适应症推广Depakote被FDA判罚15亿美元,这更让保守派对创新药业务的未来感到怀疑,因此,让股东获利并不是拆分的全部原因。
虽然在2012年时,雅培的总销售额已经达到399亿美元,几乎就是一个“青春版”的强生,但从盈利能力而言,雅培并不如强生。为了让业务获得最专业的领导,或许拆分的确是一个不错的选择。通过拆分,两个公司的活力都得到巨大的释放,不论是改革派和革新派都得到了他们想要的发展模式,最终两个公司都实现了高速累积,市盈率高速上涨。
主要的并购交易
2014年,艾伯维收购ImmuVen,价格未知
2015年,艾伯维收购Pharmacyclic,获得ibrutinib销售权,210亿美元
2016年,艾伯维收购Stemcentrx,获得Rovalpituzumabtesirine,98亿美元
2016年,收购医疗器械公司St. Jude Medical,获得多个产品,25亿美元
2016年,收购医疗器械供应商Kalila Medical,价格未知
2016年,收购诊断设备供应商Alere,获得一系列产品,53亿美元
2015年,收购Tendyne,2.5亿美元
2015年,收购医疗设备公司Cephea ValveTechnologies,价格未知
2014年,收购诊断设备公司Topera,2.5亿美元
2014年,收购医疗设备供应商CardiacTherapeutics,价格未知
2014年,收购Veropharm,获得伤口护理业务,3.02亿美元
2014年,收购CFR Pharma,获得系列药品管线,33.3亿美元
2013年,收购医疗设备公司IDEV Tech,3.01亿美元
2013年,收购医疗设备公司OptiMedica,4亿美元
2012年,收购Neuro Genetic Pharma,价格未知
2010年,收购Piramal's Healthcare,获得仿制药业务,37.2亿美元
2010年,收购诊断公司Murex,0.58亿美元
2010年,收购Facet Biotech,获得Elotuzumab,4.5亿美元
2009年,收购软件开发商STARLIMS Technologies,1.23亿美元
2009年,收购Solvay Pharma,获得Creon等系列产品,66亿美元
2009年,收购医疗设备公司Evalve,4.1亿美元
2009年,收购医疗器械公司Visiogen,4亿美元
2009年,收购医疗设备公司Medical Optics,28亿美元
2006年,收购Kos Pharma,获得Niaspan等系列产品,37亿美元
2006年,收购Guidant的器械业务,60亿美元
2004年,收购医疗器械公司Spine NextSA,0.6亿美元
2004年,收购诊断公司TheraSense,12亿美元
2003年,收购诊断公司i-STAT,3.92亿美元
2003年,收购医疗器械公司Integrated Vascular Systems,0.65亿美元
2003年,收购营养品公司Zone Perfect Nutrition,1.6亿美元
2002年,收购医疗器械公司SpinalConcepts,2.1亿美元
2002年,收购JOMED N.V.的部分业务,0.7亿美元
2002年,收购Hokuriku Seiyaku,2.92亿美元
2002年,收购Biocompatibles的心血管设备业务,2.34亿美元
2001年,收购Vysis Inc,3.55亿美元
2001年,收购Millennium pharma,2.5亿美元
2001年,收购MedNova Ltd的手术器械业务,价格未知
2000年,收购BASF的制药部门Knoll AG,获得修美乐等系列产品,69亿美元
1999年,收购葛兰素威康的麻醉品管线,价格未知
1999年,收购医疗设备公司Perclose,6.44亿美元
1997年,收购赛诺菲在美国的大输液产品线,2亿美元
1996年,收购诊断公司MediSense,8.76亿美元
药事纵横投稿须知:稿费已上调,欢迎投稿

并购抗体创新药专利到期合作
100 项与 Blas Lorente Gonzalez SL 相关的药物交易
登录后查看更多信息
100 项与 Blas Lorente Gonzalez SL 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年07月08日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
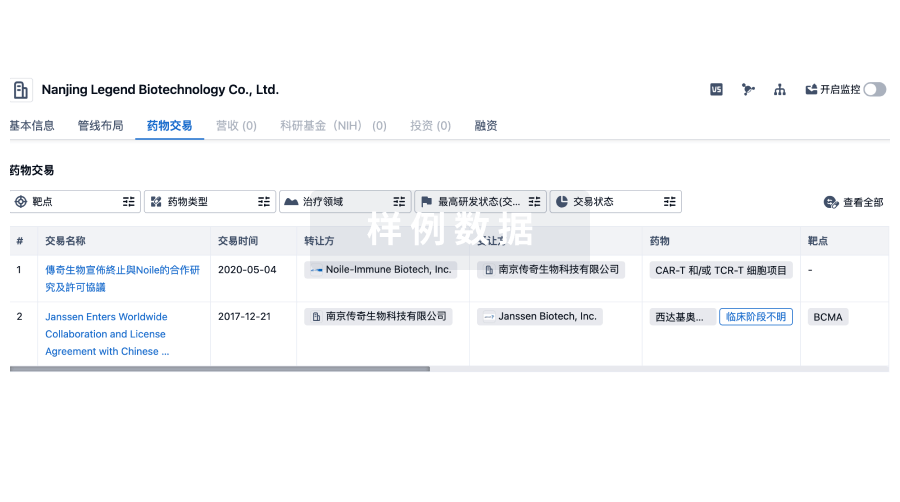
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
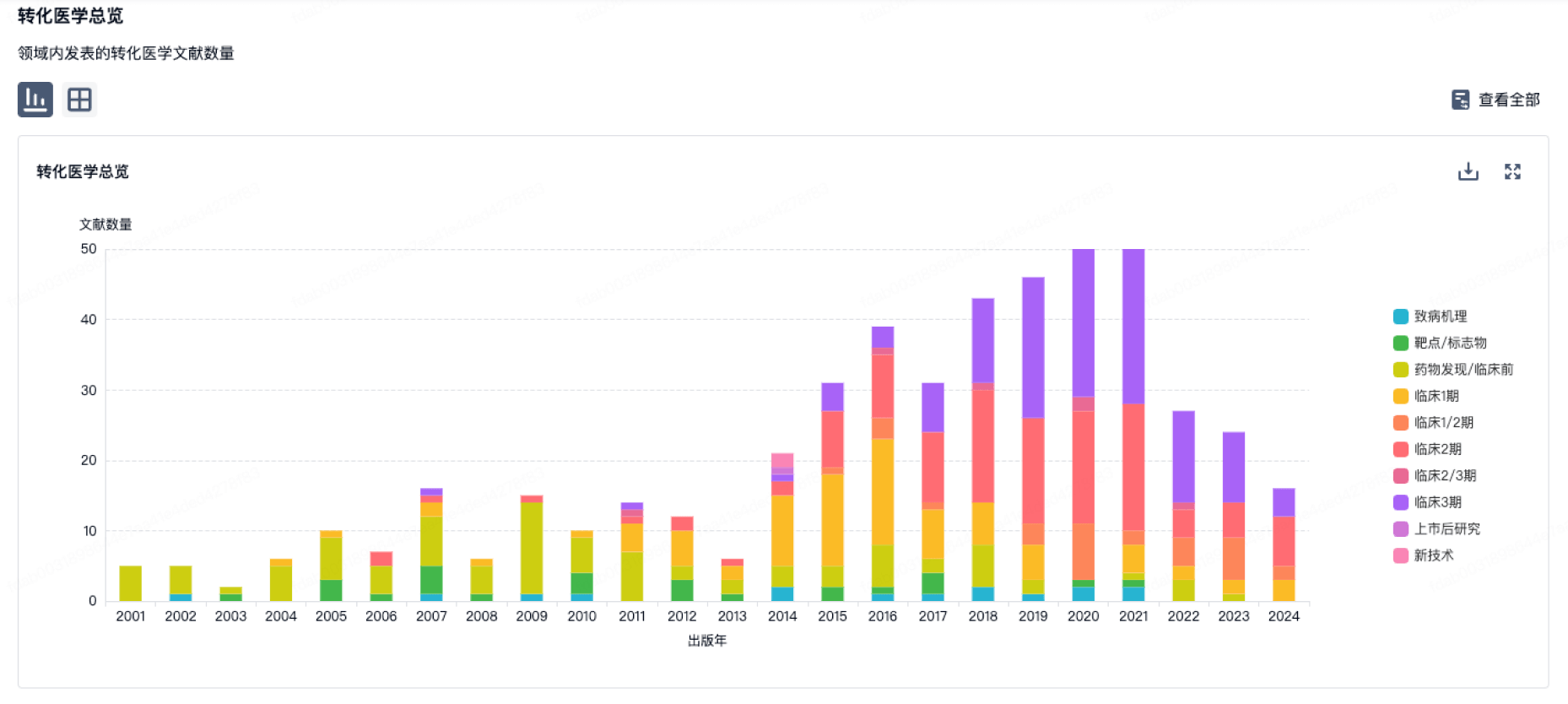
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
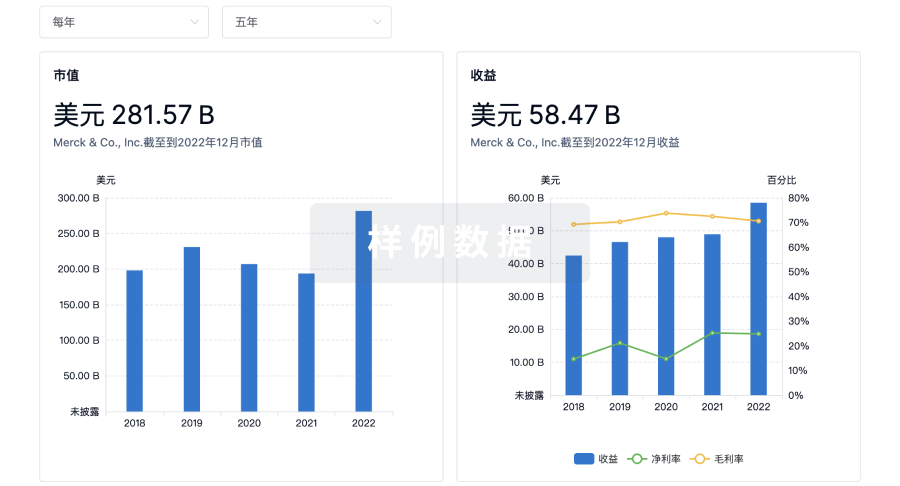
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用