更新于:2024-11-01

Gmp SA
更新于:2024-11-01
概览
关联
100 项与 Gmp SA 相关的临床结果
登录后查看更多信息
0 项与 Gmp SA 相关的专利(医药)
登录后查看更多信息
10
项与 Gmp SA 相关的新闻(医药)2024-10-13
·药通社
01 中国GMP对QA的要求
第二节质量保证
第八条质量保证是质量管理体系的一部分。企业必须建立质量保证系统,同时建立完整的文件体系,以保证系统有效运行。
第九条质量保证系统应当确保:
(一) 药品的设计与研发体现本规范的要求;
(二) 生产管理和质量控制活动符合本规范的要求;
(三)管理职责明确;
(四)采购和使用的原辅料和包装材料正确无误;
(五)中间产品得到有效控制;
(六)确认、验证的实施;
(七)严格按照规程进行生产、检查、检验和复核;
(八)每批产品经质量受权人批准后方可放行;
(九)在贮存、发运和随后的各种操作过程中有保证药品质量的适当措施;
(十)按照自检操作规程,定期检查评估质量保证系统的有效性和适用性。
第十条药品生产质量管理的基本要求:
(一)制定生产工艺,系统地回顾并证明其可持续稳定地生产出符合要求的产品;
(二)生产工艺及其重大变更均经过验证;
(三)配备所需的资源,至少包括:
1.具有适当的资质并经培训合格的人员;
2.足够的厂房和空间;
3.适用的设备和维修保障;
4.正确的原辅料、包装材料和标签;
5.经批准的工艺规程和操作规程;
6.适当的贮运条件。
(四)应当使用准确、易懂的语言制定操作规程;
(五)操作人员经过培训,能够按照操作规程正确操作;
(六)生产全过程应当有记录,偏差均经过调查并记录;
(七)批记录和发运记录应当能够追溯批产品的完整历史,并妥善保存、便于查阅;
(八)降低药品发运过程中的质量风险;
(九)建立药品召回系统,确保能够召回任何一批已发运销售的产品;
(十)调查导致药品投诉和质量缺陷的原因,并采取措施,防止类似质量缺陷再次发生。
02 ICH Q7中对质量管理的描述
QUALITY MANAGEMENT 质量管理
2.1 Principles 原则
2.10 Quality should be the responsibility of all persons involved in manufacturing.
质量应该是所有参与生产的人员的职责。
2.11 Each manufacturer should establish, document, and implement an effective system for managing quality that involves the active participation of management and appropriate manufacturing personnel.
每个生产企业应该建立、文件化并执行一个有管理层和适当的生产人员参与的有效的管理质量的体系。
2.12 The system for managing quality should encompass the organisational structure, procedures, processes and resources, as well as activities necessary to ensure confidence that the API will meet its intended specifications for quality and purity. All quality related activities should be defined and documented.
管理质量的体系应该包含组织架构、程序、流程和资源,以及确保原料药有信心符合预期的质量和纯度标准的必要的活动。所有质量相关的活动都应该规定并使其文件化。
2.13 There should be a quality unit(s) that is independent of production and that fulfills both quality assurance (QA) and quality control (QC) responsibilities. This can be in the form of separate QA and QC units or a single individual or group, depending upon the size and structure of the orgnization.
应该有一个独立于生产并且要履行质量保证(QA)和质量控制(QC)职责的质量单元(或质量部门)。根据组织的大小和架构,这个质量单元可以由单独的QA和QC单元或一个人或团队组成。
2.14 The persons authorised to release intermediates and APIs should be specified.
应当指定被授权放行中间体和原料药的人员。
2.15 All quality related activities should be recorded at the time they are performed.
所有质量相关的活动都应该在其执行的时候就记录。
2.16 Any deviation from established procedures should be documented and explained. Critical deviations should be investigated, and the investigation and its conclusions should be documented.
任何与已经建立的程序的偏离都应该记录(或登记在案并解释。
2.17 No materials should be released or used before the satisfactory completion of evaluation by the quality unit(s) unless there are appropriate systems in place to allow for such use (e.g. release under quarantine as described in Section 10.20 or the use of raw materials or intermediates pending completion of evaluation).
在质量单元(部门)圆满完成评价前物料不应该放行或使用,除非在合适的位置有合适的系统允许这样的使用(比如在第10.20章节中描述的待验状态下的放行或等待完成评价时的原料或中间体的使用)。
2.18 Procedures should exist for notifying responsible management in a timely manner of regulatory inspections, serious GMP deficiencies, product defects and related actions (e.g., quality related complaints, recalls, regulatory actions, etc.).
应该存在以一种及时的方式通知负责的管理层相关监管检查、严重的GMP缺陷、产品缺陷和相关措施的程序(比如质量相关的投诉、召回、监管行动等)。
2.2 Responsibilities of the Quality Unit(s)
质量单元的职责
2.20 The quality unit(s) should be involved in all quality-related matters.
质量单元应该参与所有质量相关的事项。
2.21 The quality unit(s) should review and approve all appropriate quality-related documents.
质量单元应该审核和批准所有质量相关的文件。
2.22 The main responsibilities of the independent quality unit(s) should not be delegated. These responsibilities should be described in writing and should include but not necessarily be limited to:
独立的质量单元的主要职责不应该被委托(给他人)。这些职责应该书面描述并应该包括但不必限于以下内容:
1. Releasing or rejecting all APIs. Releasing or rejecting intermediates for useoutside the control of the manufacturing company;
放行或拒收所有原料药。放行或拒收在生产企业控制范围之外使用的中间体;
2. Establishing a system to release or reject raw materials, intermediates,packaging and labelling materials;
建立一个放行或拒收原料、中间体、包材和标签的系统;
3. Reviewing completed batch production and laboratory control records of critical process steps before release of the API for distribution;
放行销售的原料药前审核关键工艺步骤的已完成的批生产记录和实验控制记录;
4. Making sure that critical deviations are investigated and resolved;
确保关键偏差得到调查和解决;
5. Approving all specifications and master production instructions;
批准所有标准和生产工艺规程;
6. Approving all procedures impacting the quality of intermediates or APIs;
批准所有影响中间体或原料药质量的规程;
7. Making sure that internal audits (self-inspections) are performed;
确保进行内部审计(自检);
8. Approving intermediate and API contract manufacturers;
批准中间体和原料药的合同生产商;
9. Approving changes that potentially impact intermediate or API quality;
批准潜在影响中间体或原料药质量的变更;
10. Reviewing and approving validation protocols and reports;
审核和批准验证方案和报告;
11. Making sure that quality related complaints are investigated and resolved;
确保质量相关的投诉得到调查和解决;
12. Making sure that effective systems are used for maintaining and calibratingcritical equipment;
确保有有效的系统用于维护和校验关键设备;
13. Making sure that materials are appropriately tested and the results are reported;
确保物料都得到适宜的检测并报告其结果;
14. Making sure that there is stability data to support retest or expiry dates and storage conditions on APIs and/or intermediates where appropriate;
确保在适当的情况下,有稳定性数据支持中间体或原料药的复验期或有效期和储存条件。
15. Performing product quality reviews
(as defined in Section 2.5).
进行产品质量回顾(在2.5章节中定义)
03 WHO GMP中的描述
Quality management in the drug industry
医药行业的质量管理
In the drug industry at large, quality management is usually defined as the aspect of management function that determines and implements the “quality policy”,i.e. the overall intention and direction of an organization regarding quality, as formally expressed and authorized by top management. The basic elements of quality management are:
在大的医药行业中,质量管理常常被定义为管理职能中规定并执行“质量政策”的那方面职能。比如组织质量相关的总体意图和方向,由最高管理层正式表达和授权。
质量管理的基本要素有:
— an appropriate infrastructure or “quality system”, encompassing the organizational structure, procedures, processes and resources;
一个适当的基础架构或“质量体系”,包含组织架构、程序、流程和资源;
— systematic actions necessary to ensure adequate confidence that a product (or service) will satisfy given requirements for quality. The totality of these actions is termed “quality assurance”.
确保一个产品(或服务)有足够信心会满足质量要求的不要的系统化的行动。全部这些行动被称之为“质量保证”。
Within an organization, quality assurance serves as a management tool. In contractual situations, quality assurance also serves to generate confidence in the supplier. The concepts of quality assurance, GMP and quality control are interrelated aspects of quality management. They are described here in order to emphasize their relationship and their fundamental importance to the production and control of pharmaceutical products.
在一个组织内,质量保证是一种管理工具。在委托情况下,质量保证也用于产生对供应商的信心。质量保证、GMP和质量控制的概念是质量管理的相关要素。它们在这里描述是为了强调它们的关系和它们对于药品生产和控制的根本的重要性。
1. Quality assurance 质量保证
1.1 Principle. 原则
“Quality assurance” is a wide-ranging concept covering all matters that individually or collectively influence the quality of a product. It is the totality of the arrangements made with the object of ensuring that pharmaceutical products are of the quality required for their intended use. Quality assurance therefore incorporates GMP and other factors, including those outside the scope of this guide such as product design and development.
“质量保证”是一个涵盖所有个体或集体影响产品质量的因素很宽泛的概念。它是做出的确保药品质量符合其预定用途的目标的所有安排的总和。所以质量保证整合了GMP和其他因素,包括那些不在本指南范围比如产品设计和开发内的那些因素。
1.2 The system of quality assurance appropriate to the manufacture of pharmaceutical products should ensure that:
适用于药品生产的质量保证体系应该确保:
(a) pharmaceutical products are designed and developed in a way that takes account of the requirements of GMP and other associated codes such as those of good laboratory practice (GLP) 1 and good clinical practice (GCP);
药品在一定程度上考虑GMP和其他相关法规比如GLP和GCP的要求进行设计和开发;
(b) production and control operations are clearly specified in a written form and GMP requirements are adopted;
用书面的形式明确规定生产和控制操作并采用GMP要求;
(c) managerial responsibilities are clearly specified in job descriptions;
在岗位描述中明确规定管理职责;
(d) arrangements are made for the manufacture, supply and use of the correct starting and packaging materials;
为正确的起始物料和包装材料的生产、供应和使用做出一些安排;
(e) all necessary controls on starting materials, intermediate products, and bulk products and other in-process controls, calibrations, and validations are carried out;
实施所有对起始物料、中间产品和最终产品的必要的控制和其他中间过程控制、校验和验证;
(f) the finished product is correctly processed and checked, according to the defined procedures;
按照规定的程序正确地加工和检查最终产品;
(g) pharmaceutical products are not sold or supplied before the authorized persons (see also sections 9.11 and 9.12) have certified that each production batch has been produced and controlled in accordance with the requirements of the marketing authorization and any other regulations relevant to the production, control and release of pharmaceutical products;
在被授权的人员确认每批产品已经按照上市许可和其他与药品的生产、控制和放行相关的法规生产和控制前药品不能销售或供应;
(h) satisfactory arrangements exist to ensure, as far as possible, that the pharmaceutical products are stored by the manufacturer, distributed, and subsequently handled so that quality is maintained throughout their shelf-life;
存在尽可能确保药品由生产企业储存、销售和后续处理的满意的安排以确保质量在它们整个货架期都得到保持;
(i) there is a procedure for self-inspection and/or quality audit that regularly appraises the effectiveness and applicability of the quality assurance system;
有一个定期评价质量保证体系的质量和适用性的自检和/或质量审计程序;
(j) deviations are reported, investigated and recorded;
偏差被报告、调查和记录;
(k) there is a system for approving changes that may have an impact on product quality;
有批准可能对产品质量有影响的变更;
(l) regular evaluations of the quality of pharmaceutical products should be conducted with the objective of verifying the consistency of the process and ensuring its continuous improvement.
应该执行对药品质量的定期评价,目标是确认工艺的一致性并确保其持续改进。
1.3 The manufacturer must assume responsibility for the quality of the pharmaceutical products to ensure that they are fit for their intended use, comply with the requirements of the marketing authorization and do not place patients at risk due to inadequate safety, quality or efficacy. The attainment of this quality objective is the responsibility of senior management and requires the participation and commitment of staff in many different departments and at all levels within the company, the company’s suppliers, and the distributors. To achieve the quality objective reliably there must be a comprehensively designed and correctly implemented system of quality assurance incorporating GMP and quality control. It should be fully documented and its effectiveness monitored. All parts of the quality assurance system should be adequately staffed with competent personnel, and should have suitable and sufficient premises, equipment, and facilities.
生产企业必须承担药品质量的职责以确保它们适用于其既定的用途,符合上市许可的要求并且没有因为安全性、质量和功效不足对患者产生风险。达到这个质量目标是高层管理者的职责需要公司内不同部门和所有层级的员工、公司的供应商和经销商的参与和承诺。为了可靠地达到质量目标必须有一个全面设计和正确实施的质量保证结合GMP和质量控制的体系。这个体系应该完全文件化并监控其有效性。质量保证体系的所有部分都要配备足够的有资质的人员并且应该合适和足够的厂房、设备和设施。
2. Good manufacturing practices for pharmaceutical products (GMP)
药品的GMP
2.1 Good manufacturing practice is that part of quality assurance which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization. GMP are aimed primarily at diminishing the risks inherent in any pharmaceutical production. Such risks are essentially of two types: cross contamination (in particular of unexpected contaminants) and mix-ups (confusion) caused by, for example, false labels being put on containers. Under GMP:
GMP是质量保证的一部分,确保产品可以被持续地生产和控制以符合适用其预期用途和满足上市许可的质量标准。GMP的主要目标是减少药品生产的固有风险。这些风险基本有两个类型:交叉污染(特别是非预期的污染)和混乱(混淆)例如由贴在容器上的错误标签引起的。在GMP下:
(a) all manufacturing processes are clearly defined, systematically reviewed in the light of experience, and shown to be capable of consistently manufacturing pharmaceutical products of the required quality that comply with their specifications;
所有生产工艺都要明确规定、根据经验系统审核并证明其能够有能力持续一致地生产要求符合它们标准的质量的药品;
(b) qualification and validation are performed;
进行确认和验证;
(c) all necessary resources are provided, including:
提供所有必须的资源,包括:
(i) appropriately qualified and trained personnel;
合适的有资质并经过培训的人员;(ii) adequate premises and space;
足够的厂房和空间;(iii) suitable equipment and services;
合适的设备和服务;(iv) appropriate materials, containers and labels;
合适的物料、容器和标签;
(v) approved procedures and instructions;
批准的程序和工艺规程;(vi) suitable storage and transport;
合适的储存和运输;(vii) adequate personnel, laboratories and equipment for in-processcontrols;
足够的人员、实验室和设备用于中间过程控制;
(d) instructions and procedures are written in clear and unambiguouslanguage, specifically applicable to the facilities provided;
用清晰并没有歧义的语言编写的说明书和程序
(e) operators are trained to carry out procedures correctly;
培训操作人员正确地执行程序;
(f) records are made (manually and/or by recording instruments) during manufacture to show that all the steps required by the defined procedures and instructions have in fact been taken and that the quantity and quality of the product are as expected; any significant deviations are fully recorded and investigated;
在生产中做记录(手工和/或通过记录仪)以显示所有由规定的程序和规程要求的所有步骤都已经在事实上被执行,产品的数量和质量跟期望的一样;任何重大偏差都被完整记录并调查;
(g) records covering manufacture and distribution, which enable the complete history of a batch to be traced, are retained in a comprehensible and accessible form;
以可理解和可以获取的形式保存涵盖生产和销售的可以帮助追踪到一批产品的完整的历史的记录;
(h) the proper storage and distribution of the products minimizes any risk to their quality;
正确地储存和分发产品减少对其质量的风险;
(i) a system is available to recall any batch of product from sale or supply;
可用的从销售或供应端召回任意批产品的系统;
(j) complaints about marketed products areexamined, the causes of quality defects investigated, and appropriate measures taken in respect of the defective products to prevent recurrence.
对已销售产品的投诉进行检查、调查质量缺陷的原因并采取适当措施避免缺陷产品再次出现。
在制药企业的生产过程中,变更不可避免。
当生产过程、设备或原材料等发生变更时,比如更换了一种新的辅料供应商,QA 人员需要通过变更管理流程来评估这种变更对药品质量的影响。变更往往伴随着风险。QA 人员通过学习变更管理,可以更好地识别、评估和控制变更带来的风险。他们可以利用风险评估工具,如失效模式与效应分析(FMEA),来确定变更可能导致的潜在风险点。
投稿/内容沟通:华籍美人(Ww_150525)
近期精华文章
带量采购
2024-10-09
作者:雷继峰
来自:蒲公英Ouryao
什么是仿制药?
简而言之,仿制药与原研药治疗效果相同,安全性相同。仿制药因成本较原研药低许多,其价格远低于原研药的价格。
【仿制药的定义】仿制药是与原研药(也称专利药或品牌药)具有相同活性成分,相同剂量,相同给药途径,相同剂型和相同适应症的药物。例如,“顺尔宁”普通片是美国MSD公司的原研药,“顺尔宁”与其仿制药的活性成分都是孟鲁司特钠,剂量都是10mg,给药途径都是口服,剂型都是片剂,适应症都是治疗哮喘和过敏性鼻炎。
如今的仿制药和以前的仿制药有什么区别呢?细心的朋友们可以发现最近几年上市的仿制药品,其包装盒上多了一个图案,表明该仿制药通过国家药品监督管理局“仿制药一致性评价”。
什么是“仿制药一致性评价”呢?
“仿制药一致性评价”是国家药品监督管理局(简称药监局)自2015年在全国范围内开展的一项旨在提升现有仿制药质量和疗效的一项利民利国的评价工程,“仿制药一致性评价”是对2015年前已上市获批的仿制药在质量和疗效上与原研药(也称专利药或品牌药)一致性的科学评价,对2015年以后申报的仿制药按同样的科学评价标准进行评价审评,评价审评主要包含以下几个方面:
第一,药学等效性【英文是Pharmaceutical Equivalence,简称PE】的评价,仿制药与原研药须具有相同的活性成分、相同的剂量、相同的给药途径、相同剂型和相同的适应症等,即仿制药与原研药必须五个相同。
除了以上五个相同外,仿制药的质量标准须与原研药的质量标准相当,即仿制药必须符合适用的质量标准,我国药监局药品审评部门(Center of Drug Evaluation,简称CDE)审评时,会参考原研药的质量标准,要求仿制药的质量标准不低于原研药的质量标准。
原研药若在中美欧已获批上市,我国仿制药还需要符合我国药典(Chinese Pharmacopeia)或国家药监局批准的药品质量标准、美国药典(US Pharmacopeia)和欧洲药典(EU Pharmacopeia)。
药品的质量标准包括药物活性成份(API,Active Pharmaceutical Ingredient)的质量标准和制剂成品(Finished Products)的质量标准,常用的制剂剂型以口服固体制剂如片剂和胶囊为主,其质量标准包括含量,含量均匀度,杂质,崩解,溶出度等质量指标;无菌制剂如注射剂,滴眼剂等其质量标准包括无菌性、装量、杂质、可见异物、澄明度等质量指标,且要求仿制药的辅料成分和用量须与原研药一致。
药品审评部门审评时更关注通过工艺过程保证药品质量,即关注所谓的CMC(Chemistry & Manufacture Control化学和生产控制),通过活性原料药的物理化学性质的研究和控制,通过辅料和包装材料等物料的质量控制,处方配比和工艺过程的控制,中间产品的质量控制等实现制剂成品的质量控制和保证。
第二,生物等效性【英文是Bioequivalence简称BE】的评价,生物等效性评价包含许多科学的评价方法,各国药监机构都将药代动力学(PK)方法作为首选和主要的评价方法,药代动力学方法就是测量药物活性成分在生物体液中(血浆,尿液等)的浓度,要求仿制药的药物活性成分在体内的吸收、分布和代谢情况与原研药的一致。其他BE的评价方法有:体内药效对照法(PD)、临床试验终点(Clinical End Point)、体外对照法(InVitro)方法如体外溶出度和其他物理化学指标以及药监机构认可的其它方法。
药代动力学(PK)方法是指同一组受试者(通常为健康人,个别情况下为患者等)分别服用仿制药和原研药后,测量在受试者体液中的药物浓度随时间的变化,服用仿制药和原研药后的同一组人群中具有几乎相同的药物吸收速率和程度则认为生物等效性。
具体讲,一组受试者先服用仿制药,定时取血来检测其血药浓度(血液中药物的浓度),经过一段时间清洗期后仿制药在人体内浓度几乎为零时,同一组受试者再服用原研药,同样取血来检测其血药浓度,比较同一组受试者服用仿制药和原研药后的药物在血液中的浓度和吸收的程度(科学上使用Cmax和AUC两个指标),两个指标符合预定的国际通行的接受标准,则视为生物等效。
美国,欧洲,日本和世界卫生组织在审评仿制药均非常一致地认为仿制药同时具备与原研药的药学等效和生物等效时,就是治疗等效,在临床使用上可以相互替代。
即治疗等效=药学等效+生物等效。
我国药监局在审评仿制药时的评价方法和接受标准完全与欧美日等发达国家相同。
第三,相同的适应症,仿制药与原研药(参比制剂)须具有相同的说明书,相同的使用条件和适应症。即仿制药通常与原研药在治疗疾病的种类上、用药人群、用法用量上保持一致。
第四,研发和生产企业均遵守相同的研发质量管理规范(GLP/GCP)和生产质量管理规范(GMP)。
仿制药的质量和疗效源于设计和研发,研发企业以药学等效和生物等效为目标,设计处方和工艺,制定质量标准并开展生物等效的研究。
仿制药的研发和生物等效的试验研究须符合国际通行的管理规则(例如GCP,GPLP以及ICHQ7,Q8和Q9等技术指南,ICH:International Conference of Homonization)和国家药监局CDE颁布的有关技术指南。
仿制药的技术转移按照国际通行的规则(例如ICHQ10,Q11,Q12等技术指南)的要求,要保证仿制药的生产工艺和检验技术知识转移给经过培训的工厂相关人员
仿制药的生产须符合以上ICH相关质量管理规则及国家颁布的药品生产质量管理规范简称Good Manufacture Practice,简称GMP,GMP对生产药品的从业人员的资质和职责、厂房设施和生产设备的条件,生产工艺过程,分析检验方法,质量管理体系等有明确严格的要求;保证仿制药的生产过程没有污染和交叉污染;保证仿制药的生产和检验按照CDE批准的生产工艺和分析方法进行。
第五,国家药品监督管理局(简称药监局)的药品审评部门(CDE)对上述有关“药学等效和生物等效”的研究和试验资料按照国际通行的技术标准和程序进行严格的审评;药监局药品审核查验部门还要对药品研发机构,药品临床试验机构(包括生物等效性研究机构或临床研究机构)和药品生产企业等进行现场核查,确保药品研制和生产提交的数据真实可靠,均按照GMP执行;药监局药品检验部门还要对申报的仿制药的药品质量按照原研药相同的质量标准进行复核,对仿制药的样品进行检验。
符合上述几个方面要求并在审评、核查、检验均合格后,由药品审评部门(CDE)提交建议批准,再经药监局注册管理部门(药品注册司)再次复核后,仿制药才会获得药监局的“仿制药一致性评价”的批准。
通过“仿制药一致性评价”并非容易
国内外的医生和患者都对仿制药的质量和疗效有一定程度的顾虑和担心。2015年以来,国家药监对已经上市销售的仿制药开展“仿制药一致性评价”,对新申请的仿制药严格按照一致性评价的标准要求进行审评,“一致性评价”的严格要求为患者带来药品有效性和安全性的保障。
仿制药的研发和生产是一个科学研究和技术创新的过程,原研药在处方和工艺技术有各种专利和非专利壁垒,需要逐一攻克。仿制药研发和生产并非易事,也是一个复杂,漫长又艰难的过程。
有些原研药其化合物专利过期多年了,依然没有仿制药上市,例如有些吸入制剂、外用制剂、长效缓控释注射剂等。
原研药高昂的价格限制了患者的广泛使用,各国政府从公共健康和控制医药费用两个角度考虑,都在出台各种政策鼓励仿制药的研发、生产和使用。
“仿制药一致性评价”是国家药监局基于欧美通行技术标准经过严格审评后授予的,通过一致性评价的仿制药与原研药是"治疗等效,即药学等效和生物等效的",仿制药和原研药在临床上可以相互替代。
仿制药便宜,质量疗效可靠吗?
世界各国的仿制药普遍较原研药便宜许多,老百姓普遍都认为“一分钱,一分货”,仿制药那么便宜,质量和疗效可靠吗?相信这一定是患者和临床医生的共同疑问!那么,我们来分析一下原研药的价格和仿制药的价格是如何构成?
原研药是创新的一种药物,可以理解为一种发明创造或发现创造,发明或发现药物的临床治疗作用的过程需要大量的研究资金,还需要大量资金用于长时间的临床前的安全性研究(动物试验)和临床试验研究(人体试验),用以筛选和评价药物的安全性和有效性。
新药的研发成本通常高达10亿美元,甚至50亿美元,历时十余年时间,新药一旦批准上市后,原研药公司还要雇佣大量的医药专业人才对临床医生进行新药的药理药效的教育和宣传,市场推广费用巨大。
在原研药新药专利过期后,任何制药企业均可以合法地进行仿制研究。相比之下,仿制药的研发成本较原研药的研发低了很多,仿制药不需要为临床研究投入大量资金,但需要做到与原研药的药学等效和生物等效(即上文所介绍的“一致性评价”)
综上所述,原研药的价格=药物生产成本(活性原料、辅料和生产制造费用)+新药的发明研究成本分摊+新药的临床试验成本分摊+新药的宣传和促销+较高利润,因为原研药是独此一家并有特定治疗功效,其定价是尽可能的高,只要市场可承受。而原研药的药物生产成本可能仅占原研药价格的1%-5%,除了较大的利润外,其成本主要是研发成本的分摊和巨大的市场促销费用。
仿制药的价格=药物生产成本+仿制药研发成本分摊+较小的物流和销售费用+合理利润,仿制药通常有多家制药公司,价格取决于市场竞争和生产成本,仿制药的价格通常是原研药的五分之一,十分之一,甚至更低。获得了一致性评价的仿制药与原研药的药学等效和生物等效,在临床上治疗效果一样,但价格比原研药低许多。
仿制药价格之所以低是因为省去了高昂的临床研究费用和市场推广费用,多家仿制药的竞争使其利润大大降低,但价格低的仿制药的质量和疗效等同于原研药。
疗效相同但价格低廉的仿制药使得无法负担高昂原研药的患者群体获得了用药保障,特别是慢性疾病需要长期用药的群体的用药。仿制药在我国城乡的推广使用对我国这样一个发展中国家的公共健康意义重大,对国家医保控费意义重大,对我国制药产业的质量提升和发展意义重大。
国际上,原研药专利一旦过期,经药监部门审评后就可以上市销售仿制药,仿制药的用量在临床使用上占了大多数。
美国2023年,大约有50亿个处方,其中45个亿的处方为仿制药,占处方总量的90%,而仿制药的销售额仅占10%(约600亿美元),说明仿制药在美国这样的发达国家为患者在广泛的疾病领域提供了最基本的治疗药物,美国仿制药用量巨大且价格低,仿制药为患者和政府节省了巨额支出,因此美国政府和药监局FDA非常重视仿制药的审批和使用,美国FDA主动向公众宣传仿制药,公开宣称FDA批准的仿制药其质量疗效与原研药相同,鼓励医生和患者使用仿制药(见附图1-4,FDA在各种场合宣传仿制药)。
另外,在美国每一个原研药均有其商品名,原研药公司的医药代表常年向医生推销药品的商品名,因而医生仅仅知道原研药的商品名及其治疗作用,如商品名“Singulair”是美国默沙东公司的原研药,用以治疗哮喘和过敏性鼻炎,美国医生给哮喘或过敏性鼻炎的患者开药方时,只写原研药的商品名“Singulair”的处方,医生可能并不知道其化学名是“MontelukastSodium孟鲁司特钠”。
美国患者拿到商品名的“Singulair”处方后,去连锁药店取药,在美国医院和药店是完全分离的,医院内通常没有药店,患者只能去连锁药店取药(美国有三大连锁药药店,CVS,Walgreens和RedAid),一旦有仿制药获批上市,这些连锁药店就会采购仿制药并给患者配“Singular”的仿制药。通常仿制药获得FDA批准后,仿制药就可以通过竞标进入连锁药店,连锁药店就会给患者配仿制药,因为美国各州立法要求仿制药的强制替代(Compulsory Substitution),商业保险公司也按仿制药的价格给患者报销药费。
美国仿制药的这种法规安排,其实与我国国家医保局药品带量集中采购是一样的,两国均由药品支付方负责仿制药的采购和使用。
在美国,仿制药不需要向医生推销,仿制药不需要进入医院,而是经过竞标(价格竞争,通常是独家中标)进入连锁药店或药品批发企业,其市场和销售费用较原研药低许多!
国采只选用通过“一致性评价”的仿制药
国家药品带量集中采购只选用通过“一致性评价”的仿制药,国家药监局严格监控集采中选仿制药。
为了降低我国患者的医药负担和国家医保支付负担,2018年12月新组建的国家医保局参照国际相关实践,在我国4个直辖市和7个城市率先实施药品带量集中采购,采购的对象只限于通过“一致性评价”仿制药。
获得国家药监局“一致性评价”的仿制药有资格参与国家医保局组织的“4+7”城市药品带量采购,中标的仿制药平均降价幅度52%,中标后这些过评的仿制药快速进入“4+7”城市几乎所有医院和部分医保药店,为百姓带来了有疗效可负担的好药,仿制药企业也大大降低了仿制药的市场销售费用。
2018年12月至今6年来,国家医保局已开展九批十次药品带量集中采购,共纳入374个品种,平均每批约纳入42个品种,中选仿制药平均降价超过50%,涉及金额约占公立医疗机构化学药和生物药年采购额的35%。
其中第五批及第七批最高降幅甚至超过了98%,通过推进国家药品带量采购的方式,大幅度降低了国内患者的用药负担。
集采药品涵盖抗感染、肿瘤、心脑血管疾病、胃肠道疾病、精神疾病等常见病、慢性病用药,以及用于抢救休克的多巴胺注射剂、用于催产的缩宫素注射剂等抢救药、短缺药、重点监测药品,涵盖十余个治疗领域,可以满足绝大部分临床治疗需求,满足“逐步覆盖各类药品”的要求,患者受益面进一步拓宽。
自2015年国家施行仿制药一致性评价以来,国家药监局坚定稳步推进了仿制药质量和疗效一致性的科学评价,严格按照国际通行的技术标准和方法开展仿制药的一致性评价,通过一致性评价品种已占临床常用化学药品的三分之二。
为了确保国家集采仿制药的质量稳定可控,国家药监局对集采中选药品的供应企业,实行生产企业检查和中选品种抽检两个100%全覆盖,确保中选产品“降价不降质”。
对于抽检有质量问题的仿制药企业,将列入国家集采违规名单,企业将面临取消中选资格、在2年时间内禁止参与国家及省级带量采购,从机制设计上,促使企业确保了中选集采产品的质量达到规定标准。
患者反馈与真实世界研究
在2016年后新申报批准的仿制药,其监管标准已经达到国际标准,所以视同为通过一致性评价,也给予仿制药一致性评价标志。
但还是有个别患者在服用了通过一致性的仿制药后感觉效果不如原研药,这可能是多方面的原因造成的,不能因此而否定仿制药,有个体差异,也有心理作用,总感觉几角钱的仿制药不如几元钱的原研药效果好。
针对这些疑问,2021年6月起,首都医科大学宣武医院牵头对23个集采中选的代表性品种,开展临床疗效和安全性的真实世界研究,试验结果表明这些通过一致性评价的仿制药临床疗效和安全性与原研药相当。
一致性评价上市批准
2024-10-08
·同写意
本文作者
雷继峰
上海安必生制药董事长
1
什么是仿制药?
简而言之,仿制药与原研药治疗效果相同,安全性相同。仿制药因成本较原研药低许多,其价格远低于原研药的价格。
【仿制药的定义】仿制药是与原研药(也称专利药或品牌药)具有相同活性成分,相同剂量,相同给药途径,相同剂型和相同适应症的药物。例如,“顺尔宁”普通片是美国MSD公司的原研药,“顺尔宁”与其仿制药的活性成分都是孟鲁司特钠,剂量都是10mg,给药途径都是口服,剂型都是片剂,适应症都是治疗哮喘和过敏性鼻炎。
如今的仿制药和以前的仿制药有什么区别呢?细心的朋友们可以发现最近几年上市的仿制药品,其包装盒上多了一个图案,表明该仿制药通过国家药品监督管理局“仿制药一致性评价”。
2
什么是“仿制药一致性评价”呢?
“仿制药一致性评价”是国家药品监督管理局(简称药监局)自2015年在全国范围内开展的一项旨在提升现有仿制药质量和疗效的一项利民利国的评价工程,“仿制药一致性评价”是对2015年前已上市获批的仿制药在质量和疗效上与原研药(也称专利药或品牌药)一致性的科学评价,对2015年以后申报的仿制药按同样的科学评价标准进行评价审评,评价审评主要包含以下几个方面:
第一,药学等效性【英文是Pharmaceutical Equivalence,简称PE】的评价,仿制药与原研药须具有相同的活性成分、相同的剂量、相同的给药途径、相同剂型和相同的适应症等,即仿制药与原研药必须五个相同。
除了以上五个相同外,仿制药的质量标准须与原研药的质量标准相当,即仿制药必须符合适用的质量标准,我国药监局药品审评部门(Center of Drug Evaluation,简称CDE)审评时,会参考原研药的质量标准,要求仿制药的质量标准不低于原研药的质量标准。原研药若在中美欧已获批上市,我国仿制药还需要符合我国药典(Chinese Pharmacopeia)或国家药监局批准的药品质量标准、美国药典(US Pharmacopeia)和欧洲药典(EU Pharmacopeia)。
药品的质量标准包括药物活性成份(API,Active Pharmaceutical Ingredient)的质量标准和制剂成品(Finished Products)的质量标准,常用的制剂剂型以口服固体制剂如片剂和胶囊为主,其质量标准包括含量,含量均匀度,杂质,崩解,溶出度等质量指标;无菌制剂如注射剂,滴眼剂等其质量标准包括无菌性、装量、杂质、可见异物、澄明度等质量指标,且要求仿制药的辅料成分和用量须与原研药一致。
药品审评部门审评时更关注通过工艺过程保证药品质量,即关注所谓的CMC( Chemistry &Manufacture Control化学和生产控制),通过活性原料药的物理化学性质的研究和控制,通过辅料和包装材料等物料的质量控制,处方配比和工艺过程的控制,中间产品的质量控制等实现药品的质量控制和保证。
第二,生物等效性【英文是Bioequivalence简称BE】的评价,生物等效性评价包含许多科学的评价方法,各国药监机构都将药代动力学(PK)方法作为首选和主要的评价方法,药代动力学方法就是测量药物活性成分在生物体液中(血浆,尿液等)的浓度,要求仿制药的药物活性成分在体内的吸收、分布和代谢情况与原研药的一致。其他BE的评价方法有:体内药效对照法(PD)、临床试验终点法(Clinical End Point)、体外对照法(In Vitro)方法如体外溶出度和其他物理化学指标以及药监机构认可的其它方法。
药代动力学(PK)方法是指同一组受试者(通常为健康人,个别情况下为患者等)分别服用仿制药和原研药后,测量在受试者体液中的药物浓度随时间的变化,服用仿制药和原研药后的同一组人群中具有几乎相同的药物吸收速率和程度则认为生物等效。
具体讲,一组受试者先服用仿制药,定时取血来检测其血药浓度(血液中药物的浓度),经过一段时间清洗期后仿制药在人体内浓度几乎为零时,同一组受试者再服用原研药,同样取血来检测其血药浓度,比较同一组受试者服用仿制药和原研药后的药物在血液中的浓度和吸收的程度(科学上使用Cmax和AUC两个指标),两个指标符合预定的国际通行的接受标准,则视为生物等效。
美国,欧洲,日本和世界卫生组织在审评仿制药均非常一致地认为仿制药同时具备与原研药的药学等效和生物等效时,就是治疗等效,在临床使用上可以相互替代。即治疗等效=药学等效+生物等效。我国药监局在审评仿制药时的评价方法和接受标准完全与欧美日等发达国家相同。
第三,相同的适应症,仿制药与原研药(参比制剂)须具有相同的说明书,相同的使用条件和适应症。即仿制药通常与原研药在治疗疾病的种类上、用药人群、用法用量上保持一致。
第四,研发和生产企业均遵守相同的研发质量管理规范(GLP/GCP)和生产质量管理规范(GMP)。
仿制药的质量和疗效源于设计和研发,研发企业以药学等效和生物等效为目标,设计处方和工艺,制定质量标准并开展生物等效的研究。
仿制药的研发和生物等效的试验研究须符合国际通行的管理规则(例如GCP,GPLP以及ICH Q7,Q8和Q9等技术指南,ICH:International Conference of Homonization)和国家药监局CDE颁布的有关技术指南。
仿制药的技术转移按照国际通行的规则(例如ICH Q10,Q11,Q12等技术指南)的要求,要保证仿制药的生产工艺和检验技术知识转移给经过培训的工厂相关人员。
仿制药的生产须符合以上ICH相关质量管理规则及国家颁布的药品生产质量管理规范简称Good Manufacture Practice,简称GMP,GMP对生产药品的从业人员的资质和职责、厂房设施和生产设备的条件,生产工艺过程,分析检验方法,质量管理体系等有明确严格的要求;保证仿制药的生产过程没有污染和交叉污染;保证仿制药的生产和检验按照CDE批准的生产工艺和分析方法进行。
第五,国家药品监督管理局(简称药监局)的药品审评部门(CDE)对上述有关“药学等效和生物等效”的研究和试验资料按照国际通行的技术标准和程序进行严格的审评;药监局药品审核查验部门还要对药品研发机构,药品临床试验机构(包括生物等效性研究机构或临床研究机构)和药品生产企业等进行现场核查,确保药品研制和生产提交的数据真实可靠,均按照GMP执行;药监局药品检验部门还要对申报的仿制药的药品质量按照原研药相同的质量标准进行复核,对仿制药的样品进行检验。符合上述几个方面要求并在审评、核查、检验均合格后,由药品审评部门(CDE)提交建议批准,再经药监局注册管理部门(药品注册司)再次复核后,仿制药才会获得药监局的“仿制药一致性评价”的批准。
3
通过“仿制药一致性评价”并非容易
国内外的医生和患者都对仿制药的质量和疗效有一定程度的顾虑和担心。2015年以来,国家药监对已经上市销售的仿制药开展“仿制药一致性评价”,对新申请的仿制药严格按照一致性评价的标准要求进行审评,“一致性评价”的严格要求为患者带来药品有效性和安全性的保障。
仿制药的研发和生产是一个科学研究和技术创新的过程,原研药在处方和工艺技术有各种专利和非专利壁垒,需要逐一攻克。仿制药研发和生产并非易事,也是一个复杂,漫长又艰难的过程。有些原研药其化合物专利过期多年了,依然没有仿制药上市,例如有些吸入制剂、外用制剂、长效缓控释注射剂等。
原研药高昂的价格限制了患者的广泛使用,各国政府从公共健康和控制医药费用两个角度考虑,都在出台各种政策鼓励仿制药的研发、生产和使用。“仿制药一致性评价”是国家药监局基于欧美通行技术标准经过严格审评后授予的,通过一致性评价的仿制药与原研药是"治疗等效,即药学等效和生物等效的",仿制药和原研药在临床上可以相互替代。
4
仿制药便宜,质量疗效可靠吗?
世界各国的仿制药普遍较原研药便宜许多,老百姓普遍都认为“一分钱,一分货”,仿制药那么便宜,质量和疗效可靠吗?相信这一定是患者和临床医生的共同疑问!那么,我们来分析一下原研药的价格和仿制药的价格是如何构成?
原研药是创新的一种药物,可以理解为一种发明创造或发现创造,发明或发现药物的临床治疗作用的过程需要大量的研究资金,还需要大量资金用于长时间的临床前的安全性研究(动物试验)和临床试验研究(人体试验),用以筛选和评价药物的安全性和有效性。新药的研发成本通常高达10亿美元,甚至50亿美元,历时十余年时间,新药一旦批准上市后,原研药公司还要雇佣大量的医药专业人才对临床医生进行新药的药理药效的教育和宣传,市场推广费用巨大。
在原研药新药专利过期后,任何制药企业均可以合法地进行仿制研究。相比之下,仿制药的研发成本较原研药的研发低了很多,仿制药不需要为临床研究投入大量资金,但需要做到与原研药的药学等效和生物等效(即上文所介绍的“一致性评价”)。
综上所述,原研药的价格=药物生产成本(活性原料、辅料和生产制造费用)+新药的发明研究成本分摊+新药的临床试验成本分摊+新药的宣传和促销+较高利润,因为原研药是独此一家并有特定治疗功效,其定价是尽可能的高,只要市场可承受。而原研药的药物生产成本可能仅占原研药价格的1%-5%,除了较大的利润外,其成本主要是研发成本的分摊和巨大的市场促销费用。
仿制药的价格=药物生产成本+仿制药研发成本分摊+较小的物流和销售费用+合理利润,仿制药通常有多家制药公司,价格取决于市场竞争和生产成本,仿制药的价格通常是原研药的五分之一,十分之一,甚至更低。获得了一致性评价的仿制药与原研药的药学等效和生物等效,在临床上治疗效果一样,但价格比原研药低许多。仿制药价格之所以低是因为省去了高昂的临床研究费用和市场推广费用,多家仿制药的竞争使其利润大大降低,但价格低的仿制药的质量和疗效等同于原研药。
疗效相同但价格低廉的仿制药使得无法负担高昂原研药的患者群体获得了用药保障,特别是慢性疾病需要长期用药的群体的用药。仿制药在我国城乡的推广使用对我国这样一个发展中国家的公共健康意义重大,对国家医保控费意义重大,对我国制药产业的质量提升和发展意义重大。
国际上,原研药专利一旦过期,经药监部门审评后就可以上市销售仿制药,仿制药的用量在临床使用上占了大多数。美国2023年,大约有50亿个处方,其中45个亿的处方为仿制药,占处方总量的90%,而仿制药的销售额仅占10%(约600亿美元),说明仿制药在美国这样的发达国家为患者在广泛的疾病领域提供了最基本的治疗药物,美国仿制药用量巨大且价格低,仿制药为患者和政府节省了巨额支出,因此美国政府和药监局FDA非常重视仿制药的审批和使用,美国FDA主动向公众宣传仿制药,公开宣称FDA批准的仿制药其质量疗效与原研药相同,鼓励医生和患者使用仿制药(见附图1-4,FDA在各种场合宣传仿制药)。
另外,在美国每一个原研药均有其商品名,原研药公司的医药代表常年向医生推销药品的商品名,因而医生仅仅知道原研药的商品名及其治疗作用,如商品名“Singulair”是美国默沙东公司的原研药,用以治疗哮喘和过敏性鼻炎,美国医生给哮喘或过敏性鼻炎的患者开药方时,只写原研药的商品名“Singulair”的处方,医生可能并不知道其化学名是“Montelukast Sodium孟鲁司特钠”。
美国患者拿到商品名“Singulair”处方后,去连锁药店取药,在美国医院和药店是完全分离的,医院内通常没有药店,患者只能去连锁药店取药(美国有三大连锁药药店,CVS,Walgreens和RedAid),一旦有仿制药获批上市,这些连锁药店就会采购仿制药并给患者配“Singular”的仿制药。通常仿制药获得FDA批准后,仿制药就可以通过竞标进入连锁药店,连锁药店就会给患者配仿制药,因为美国各州立法要求仿制药的强制替代(Compulsory Substitution),商业保险公司也按仿制药的价格给患者报销药费。
美国仿制药的这种法规安排,其实与我国国家医保局药品带量集中采购是一样的,两国均由药品支付方负责仿制药的采购和使用。
在美国,仿制药不需要向医生推销,仿制药不需要进入医院,而是经过竞标(价格竞争,通常是独家中标)进入连锁药店或药品批发企业,其市场和销售费用较原研药低许多!
5
国家药品带量集中采购只选用通过“一致性评价”的仿制药,国家药监局严格监控集采中选仿制药
为了降低我国患者的医药负担和国家医保支付负担,2018年12月新组建的国家医保局参照国际相关实践,在我国4个直辖市和7个城市率先实施药品带量集中采购,采购的对象只限于通过“一致性评价”仿制药。获得国家药监局“一致性评价”的仿制药有资格参与国家医保局组织的“4+7”城市药品带量采购,中标的仿制药平均降价幅度52%,中标后这些过评的仿制药快速进入“4+7”城市几乎所有医院和部分医保药店,为百姓带来了有疗效可负担的好药,仿制药企业也大大降低了仿制药的市场销售费用。
2018年12月至今6年来,国家医保局已开展九批十次药品带量集中采购,共纳入374个品种,平均每批约纳入42个品种,中选仿制药平均降价超过50%,涉及金额约占公立医疗机构化学药和生物药年采购额的35%。其中第五批及第七批最高降幅甚至超过了98%,通过推进国家药品带量采购的方式,大幅度降低了国内患者的用药负担。
集采药品涵盖抗感染、肿瘤、心脑血管疾病、胃肠道疾病、精神疾病等常见病、慢性病用药,以及用于抢救休克的多巴胺注射剂、用于催产的缩宫素注射剂等抢救药、短缺药、重点监测药品,涵盖十余个治疗领域,可以满足绝大部分临床治疗需求,满足“逐步覆盖各类药品”的要求,患者受益面进一步拓宽。
自2015年国家施行仿制药一致性评价以来,国家药监局坚定稳步推进了仿制药质量和疗效一致性的科学评价,严格按照国际通行的技术标准和方法开展仿制药的一致性评价,通过一致性评价品种已占临床常用化学药品的三分之二。为了确保国家集采仿制药的质量稳定可控,国家药监局对集采中选药品的供应企业,实行生产企业检查和中选品种抽检两个100%全覆盖,确保中选产品“降价不降质”。对于抽检有质量问题的仿制药企业,将列入国家集采违规名单,企业将面临取消中选资格、在2年时间内禁止参与国家及省级带量采购,从机制设计上,促使企业确保了中选集采产品的质量达到规定标准。
6
患者反馈与真实世界研究
在2016年后新申报批准的仿制药,其监管标准已经达到国际标准,所以视同为通过一致性评价,也给予仿制药一致性评价标志。但还是有个别患者在服用了通过一致性的仿制药后感觉效果不如原研药,这可能是多方面的原因造成的,不能因此而否定仿制药,有个体差异,也有心理作用,总感觉几角钱的仿制药不如几元钱的原研药效果好。
针对这些疑问,2021年6月起,首都医科大学宣武医院牵头对23个集采中选的代表性品种,开展临床疗效和安全性的真实世界研究,试验结果表明这些通过一致性评价的仿制药临床疗效和安全性与原研药相当。
同写意媒体矩阵,欢迎关注↓↓↓
一致性评价上市批准
100 项与 Gmp SA 相关的药物交易
登录后查看更多信息
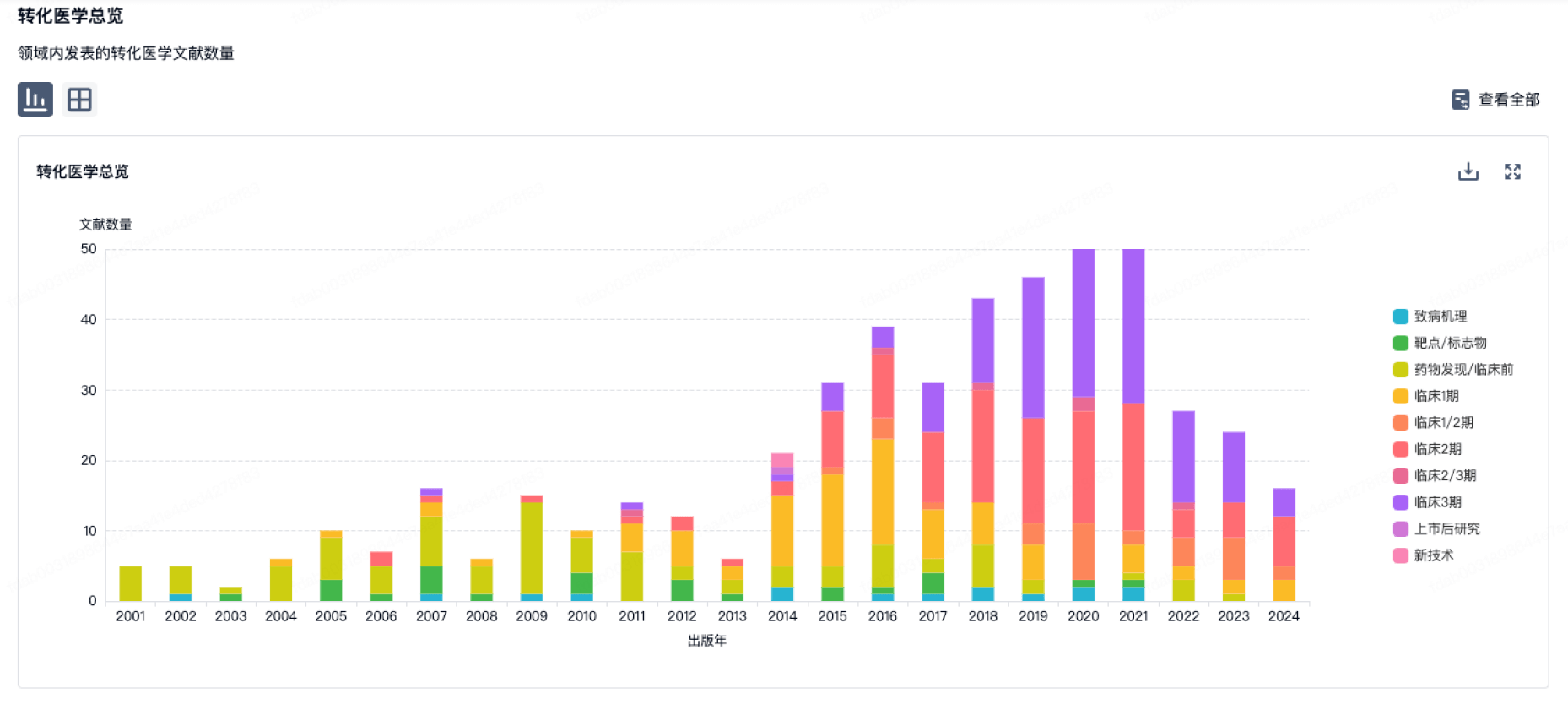
100 项与 Gmp SA 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年11月20日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或

转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
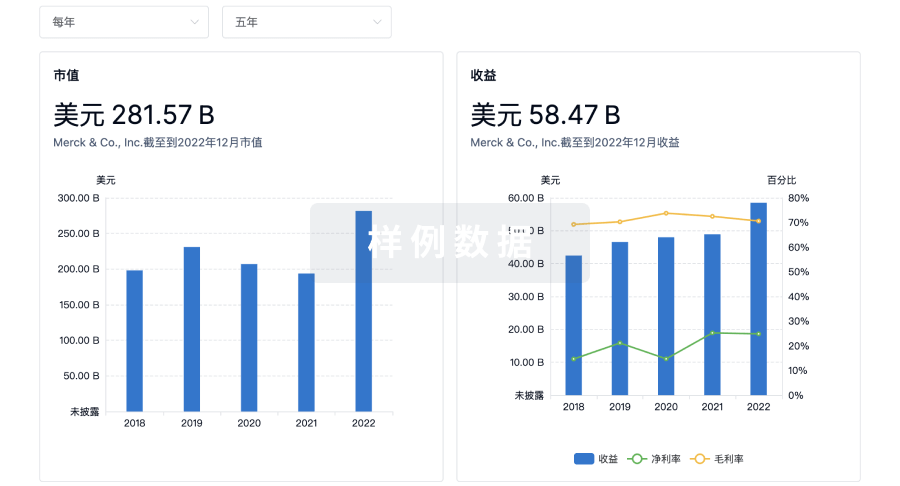
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或

投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或

融资
发掘融资趋势以验证和推进您的投资机会。
登录
或

标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用