更新于:2024-09-19
DMD PHARMACEUTICALS
更新于:2024-09-19
概览
关联
100 项与 DMD PHARMACEUTICALS 相关的临床结果
登录后查看更多信息
0 项与 DMD PHARMACEUTICALS 相关的专利(医药)
登录后查看更多信息
5
项与 DMD PHARMACEUTICALS 相关的新闻(医药)2023-10-31
前情提要y2023年6月,FDA加速批准AAV基因疗法Elevidys上市,用于治疗4至5岁杜氏肌营养不良症(DMD)儿童患者;是首款获得美国FDA批准上市的DMD基因治疗。Sarepta公司意图扩大Elevidys的批准范围,使其能够覆盖更大年龄的DMD患者。但是DMD是一种进行的疾病,患者年龄越大疾病越发进展,失去更多功能,治疗效果可能不如年纪小的患者。今日,Sarepta Therapeutics宣布了DMD基因治疗Elevidys的全球III期研究EMBARK(Study 9001-301)的顶线结果,EMBARK是一项针对4-7岁杜氏肌营养不良症(DMD)的临床研究。结果报告,Elevidys在研究中未达到主要终点。尽管EMBARK研究未达到主要终点,但是该研究数据仍然支持Elevidys向FDA提交疗效补充申请(efficacy supplement to the BLA)。值得注意的是,FDA已经表示愿意根据EMBARK研究提交的数据进行标签扩展的审查。积极的次要终点EMBARK研究的主要终点是治疗后第52周的NSAA(North Star Ambulatory Assessment)评分与基线相比的变化;Elevidys组的NSAA评分提交2.6分,安慰剂组提交1.9分;两组相差0.65分无统计学意义。但是EMBARK研究的关键次要终点,包括Time to rise (TTR)测试、10米步行测试等,在所有年龄组中都显示出具有临床意义的治疗益处。此外,其他次要终点,例如including stride velocity 95th centile (SV95C) 和 time to ascend 4 steps均显示出治疗益处。安全性方面,没有观察到新的安全信号,7名患者(11.1%)经历了与治疗相关的严重不良事件。EMBARK研究的全部结果将在之后的医学会议上分享,并在医学杂志上发表。小结通常来说,如果临床研究未达到主要终点,FDA是不会认为达到次要终点的数据是积极的。但在严重疾病或没有其他治疗选择的疾病上,FDA会采取不同的措施,例如DMD。延伸阅读:DMD基因治疗进行首位商业患者给药参考资料:1.https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-topline-results-embark-global-02.https://www.biopharmadive.com/news/sarepta-duchnne-embark-study-failure-gene-therapy-elevidys/698243/本周好文推荐如需转载请联系佰傲谷并在醒目位置注明出处﹀ ···
基因疗法临床3期加速审批临床结果
2023-08-16
·药研发
「 本文共:15条资讯,阅读时长约:3分钟 」今日头条徐诺HDAC抑制剂胶质瘤Ⅰ期临床积极。徐诺药业宣布其口服泛HDAC抑制剂艾贝司他与替莫唑胺的联合方案,在美国针对复发性胶质瘤(GBM)的Ⅰ期临床已完成首个剂量组给药和初步安全性评估,下一步将开始更高剂量组的患者入组。艾贝司他是徐诺药业的首款候选药物,此前已获得FDA授予两项快速通道资格,单药4线治疗复发/难治滤泡性淋巴瘤,以及与培唑帕尼联用治疗肾细胞癌经治患者。国内药讯1.华毅乐健血友病基因疗法完成IIT研究入组。华毅乐健自主研发的AAV基因疗法 GS1191-0445注射液在研究者发起的IIT研究完成全部12例患者入组,拟评估单次静脉输注在≥18岁且内源性凝血因子Ⅷ(FⅧ)活性<1IU/dL(即<1%)的血友病A患者中的安全性和耐受性。GS1191-0445旨在通过一次注射治疗,恢复肝细胞持续表达FVIII的功能,从而解决患者长期接受预防性凝血因子输注的需求。2.上海昂阔CDH6-ADC获批实体瘤临床。昂阔医药靶向cadherin-6(CDH6)抗体偶联药物(ADC)CUSP06获FDA批准开展Ⅰ期临床,拟评估用于治疗铂类难治/耐药卵巢癌和其他晚期实体瘤患者中的安全性和耐受性,以及初步抗肿瘤疗效。CUSP06使用的连接子专门为依沙替康载荷设计,可生成高度稳定和均质的ADC。在临床前研究中,这种连接子/有效载荷显示出较强的旁观者效应,CUSP06的药抗比为8。3.正大天晴MEK1/2抑制剂获批NF1临床。正大天晴1类化药TQ-B3234胶囊获国家药监局临床试验默示许可,临床适应症为I型神经纤维瘤病(NF1)的治疗。TQ-B3234是一种选择性MEK1/2抑制剂,主要通过对MEK蛋白的抑制作用,影响MAPK通路,抑制细胞增殖。目前,该新药正在Ⅰ期临床中评估用于中国I型神经纤维瘤病相关(神经纤维瘤和外周恶性神经鞘膜瘤)受试者中的安全性、药代动力学特征以及初步疗效。4.科望医药SIRPα单抗报IND。科望医药SIRPα阻断性抗体新药ES004注射液的临床试验申请获CDE受理。ES004可有效阻断CD47-SIRPα相互作用以及CD47介导的SIRPα胞内段对SHP-1磷酸酶的募集。通过阻断CD47介导的“抗吞噬”信号,ES004可以大大增强巨噬细胞针对肿瘤抗原特异性抗体调理后的肿瘤细胞的吞噬。在小鼠体内模型中,ES004联合Claudin18.2抗体治疗显示出积极的抗肿瘤活性,且安全性良好。5.传奇BCMA CAR-T疗法2023Q2大卖1.17亿美元。8月15日,传奇生物发布2023年二季度财务报表,上半年营收1.1亿美元,同比增长77%,研发投入1.81亿美元,同比增长20%。2023Q2,BCMA CAR-T疗法Carvykti销售额1.17亿美元,同比增长388%。今年6月,Carvykti已向FDA递交2-4线治疗多发性骨髓瘤(MM)的上市申请,PDUFA日期为2024年4月5日。国际药讯1.创新癌症肝转移治疗系统获批上市。Delcath Systems公司组合产品Hepzato系统(melphalan/肝脏输送系统)获FDA批准上市,通过肝脏输送系统直接将melphalan输送到肝脏,用于转移性葡萄膜恶性黑色素瘤(mUM)成人患者的肝导向治疗。在Ⅲ期临床中,独立审查委员会评估的客观缓解率达到36.3%,中位缓解持续时间为14个月,疾病控制率(DCR)为73.6%。2.创新抗生素组合获FDA优先审评资格。Venatorx公司β-内酰胺酶抑制剂他尼硼巴坦的新药上市申请获FDA授予优先审评资格,联合头孢吡肟治疗复杂性尿路感染(包括急性肾盂肾炎)。在Ⅲ期临床CERTAIN-1中,与美罗培南相比,该组合达到临床和微生物学应答的患者比例具有优效性(70.0%vs58%)。云顶新耀拥有该组合疗法的大中华区开发权益。3.抗体寡核苷酸偶联物获DMD孤儿药认定。Avidity Biosciences公司抗体寡核苷酸偶联物(AOC)AOC 1044获FDA授予孤儿药资格,用于治疗44号外显子跳跃突变的杜氏肌营养不良症(DMD)。AOC 1044旨在将PMO递送至骨骼肌和心脏组织,通过特异性跃过外显子44,促使患有DMD44病人产生部分功能性抗肌萎缩蛋白。2023年4月,该疗法已获FDA快速通道指定。4.腓骨肌萎缩症小核酸药物获孤儿药资格。DTx Pharma公司创新FALCON siRNA疗法DTx-1252获FDA授予孤儿药资格,用于治疗1A型腓骨肌萎缩症(CMT1A)。这是一种由施旺细胞中PMP22基因的过度表达导致的进行性神经肌肉常染色体显性遗传疾病,可导致终身肌肉功能丧失和残疾。临床前研究显示,DTx-1252能够通过抑制PMP22,诱导轴突髓鞘再形成至正常水平,改善相关电生理结果,具有逆转CMT1A的潜力。5.鼻腔给药CD3抗体获批AD临床。Tiziana公司全人源CD3单抗foralumab获FDA临床许可,拟采用鼻腔局部给药,用于治疗阿尔兹海默症。foralumab主要通过抑制大脑中小胶质细胞激活起作用,可以针对阿尔茨海默病的根本病理,解决神经炎症问题。预计将于2024年启动临床。此前,在治疗非活动性继发进展型多发性硬化(SPMS)患者的临床研究中,3个月内观察到5例患者小胶质细胞活化减少。6.AAV基因疗法公司Taysha融资10.9亿元。Taysha Gene公司宣布完成1.5亿美元(约10.9亿人民币)融资,以用于推进其自体补充鞘内递送AAV9基因转移疗法用于Rett综合征的临床开发。Ⅰ/Ⅱ期临床(REVEAL)6周治疗数据显示,TSHA-102改善了患者的疗效指标(临床整体印象量表-改善(CGI-I),临床总体印象-严重程度(CGI-S)和Rett综合征行为问卷(RSBQ)),且耐受性良好。此前,TSHA-102已获得FDA的孤儿药和罕见儿科疾病指定,以及EMA孤儿药资格。医药热点。1.感染性眼病研究中心成立。近日,由国家传染病医学中心(北京)、北京地坛医院与北京同仁医院携手共建的“国家传染病医学中心——感染性眼病研究中心”正式成立。此次合作,北京地坛医院作为主体医院,与北京同仁医院共同助力感染性眼病领域的深入研究。该中心将充分发挥双方在学科领域的优势,聚焦眼科感染领域,提升感染性眼病的临床科研核心竞争力。2.杨健教授全职加入西湖大学。近日,美国宾州州立大学杨健教授正式加入西湖大学,任生物材料及再生工程讲席教授、校长助理。杨健教授专长于可降解生物材料的开发研究及应用,是国际知名生物材料专家。杨健教授开发的生物材料具有广阔的应用前景,目前的主要研究应用方向包括疾病的诊断及治疗、生物传感器、药物释放、医疗器械,及复杂人体组织再生及创伤修复等领域。 3.周末睡懒觉或损害肠道健康。英国伦敦国王学院研究人员最新发表在《欧洲营养杂志》上的论文显示,不规律的睡眠时间与肠道健康的负面影响之间存在联系。比如工作日按照闹钟早起,周末睡到自然醒,也会影响生物节律和新陈代谢,甚至可能对肠道细菌种类产生负面影响。评审动态 1. CDE新药受理情况(08月16日) 2. FDA新药获批情况(北美08月15日)股市资讯上个交易日 A 股医药板块 -0.86%涨幅前三 跌幅前三易瑞生物+20.00% 圣诺生物 -9.18%博拓生物 +9.94% 太极集团 -7.67%康芝药业 +6.16% 宏源药业 -6.47% 【微芯生物】全资子公司成都微芯药业有限公司的产品CS32582胶囊”收到国家药监局签发的境内生产药品注册临床试验的《受理通知书》。【康希诺】公司的产品重组新型状病毒疫苗(5型腺病毒载体)克威莎®”(以下简称“克威莎)收到印度尼西亚(以下简称“印尼”)乌拉玛委员会食品、药品及化妆品评估机构的清真认证。【兴齐眼药】公司的产品“环孢素滴眼液(11)在厦门大学附属厦门眼科中心完成首例受试者入组,正式进入IV期临床试验。- The End -戳“阅读原文”,了解更多医药研发及股市资讯。
免疫疗法抗体药物偶联物快速通道临床1期细胞疗法
2023-07-16
·药时代
药时代:谁将荣登《中国制药江湖封神榜》?正文共: 6998字 43图预计阅读时间: 18分钟药时代编者按:2023年进入了7月份,各种各样的行业资讯、重磅消息纷至沓来,让人目不暇接。这些资讯、消息中很多和美国食品药品监督管理局(FDA)相关,值得中国药企、CDMO公司特别关注。现在,药时代团队挑选了其中的18条,做一个简单的汇总,供广大企业和业界同药们特别了解。这些资讯既包括FDA批准的新药、拒绝批准的新药,也包括FDA关于ChatGPT、外部专家咨询委员会(Adcomm)、细胞和基因疗法(CGT)指导文件草案、伴随诊断、现场核查等影响全球制药行业和企业的重大事项。另外,美国从中国进口化疗药物也是一个值得中国致力于出海的药企格外关注的大事。时间、水平、篇幅有限,错误疏漏难免,欢迎大家批评指正!0120年里首款获得FDA完全批准的AD新药!2023年7月6日,FDA批准了阿尔茨海默病(AD)新药LEQEMBI® (lecanemab-irmb) 100 mg/mL注射液的补充生物制剂许可申请 (SBLA),使LEQEMBI成为20年里全球首个也是唯一一个被完全批准的AD疗法,用于降低成人阿尔茨海默病(AD) 患者的疾病进展速度,延缓认知和功能衰退。(图片来源:卫材中国官微)与此相呼应,美国医疗保险和医疗补助服务中心(CMS)宣布扩大Leqembi的医疗保险范围。推荐阅读:FDA正式批准Leqembi上市02FDA批准首款非处方避孕药,预计2024年上市7月13日,Perrigo Company宣布,FDA已批准该公司的避孕药Opill由处方转换为非处方使用。这是美国首个无需处方就能获得的日常避孕药,为消费者在药店、便利店和网上轻松购买避孕药扫清了道路。Opill是一款FDA于1973年批准上市的仅含孕激素(含有0.075mg高诺酮)的每日口服避孕药,可用于所有年龄段的非处方使用。Opill最初由总部位于法国巴黎的HRA Pharma研发,2022年4月Perrigo以18亿英镑收购HRA Pharma,将这款产品收入囊中。(图片来源:Perrigo官网)值得一提的是,在FDA最新批准之前,Opill在其它国家,比如英国,就已经可以无需处方进行购买和使用了。03首款血友病A基因疗法获批,定价290万美元6月30日,主打罕见病新药研发的BioMarin Pharmaceutical宣布,美国FDA批准其基因疗法Roctavian(valoctocogene roxaparvovec)上市,用以治疗严重血友病A患者(凝血因子VIII [FVIII]活性< 1 IU/dL),其中患者体内经获FDA批准的检测确认不带有抗腺相关病毒5(AAV5)的抗体。根据新闻稿,Roctavian是首个获FDA批准治疗严重血友病A患者的基因疗法。FDA的这一批准为BioMarin曲折的新药研发和上市之路画上了句号,曾经被延迟,曾经被拒绝,现在公司终于可以转向,将这种具有重大意义的一次性疗法商业化。据透露,该一针见效的新疗法的定价为290万美元。推荐阅读:辉瑞B型血友病基因疗法3期数据积极,挑战全球最贵药物Hemgenix!史上最贵药易主!AAV基因疗法捷报成双,血友病治疗实现重大突破从280万到300万,全球最贵药物记录被打破!只用了短短30天!全球首款!血友病A“周制剂”获批上市!04关于火热的ChatGPTChatGPT为代表的大型语言模型的大量应用已经像狂风助力下的野火一样蔓延开来,势头凶猛,令全球震撼。这些走进寻常百姓家的AIGC工具让越来越多的制药界人士提出一个共同的问题,那就是:监管机构会利用这些最新的工具加快新药审批审批工作吗?近日,FDA和欧洲药品管理局(European Medicines Agency)的两位首席医学官均表示,他们不打算短期内开始使用这项新技术来加快各自机构的药品审评审批工作。这个观点出乎我们的预料,因为在我们的印象中,FDA一直鼓励创新、支持创新、倡导创新、拥抱创新。因此,禁不住要问:真的是这样吗?为什么不呢?这个短期究竟有多长呢?NMPA的态度是怎样的呢?药时代将持续关注。05FDA外部专家会议将被取消?美国FDA的外部专家咨询会制度是药品审评审批方面中非常重要的一环,随着新药研发和审评日益复杂,新的研究表明,近年来咨询委员会的数量有所下降,一位FDA专员也坚持认为,在未来的会议上,投票人数可能会减少。FDA正在探索如何改革其咨询委员会,以确保该机构获得更及时、更合理的建议。FDA承认这一过程正在进行中,而且并不容易。根据哈佛大学监管、治疗和法律项目(PORTAL)研究人员发表在《JAMA卫生论坛》上的一项分析,从2010年到2021年,FDA召开了409场与人用药相关的专家会,但“随着时间的推移,召开会议的频率越来越低,从2012年最高的50次降至2020年和2021年最低的18次。”分析指出“大部分减少涉及首次批准投票的专家会,这类会议从2012年最高的26次下降到2021年最低的8次。”FDA外部专家咨询委员会退出历史的舞台的可能性大吗?06细胞基因疗法(CGT)行业和企业注意啦!(图片来源:FDA 官网)FDA于7月13日发布了两份与细胞和基因疗法(CGT)生产有关的新指导文件草案,其中一份指示制造商在改变任何生产做法之前进行充分的风险评估,另一份则是关于如果申办方不遵守FDA规定的上市后研究将会发生什么。《人类细胞和基因治疗产品的生产变更和可比性》指南草案,提供了FDA对基于生命周期方法的细胞和基因治疗产品生产变更的管理和报告以及评估生产变更对产品质量影响的可比性研究的当前思考。由于CGT产品的复杂性,对生产变更的管理带来了许多挑战,FDA通过该指南草案为 CGT产品的研究性新药申请人(IND)和生物制品许可申请(BLA)申请人提供了有关产品可比性以及生产变更管理的建议,同时考虑适用于这些产品的独特挑战。该指南草案是在FDA将之前的组织和先进治疗办公室更名为治疗产品办公室(OTP)并将其地位提升为超级办公室之后发布的。在获得新的PDUFA VII资金后,FDA寻求利用新资金提高该办公室的审评能力,并增强其在新细胞和基因疗法方面的专业知识。07伴随诊断的监管将面临新一轮的改革?关于伴随诊断,FDA认识到一些新的抗癌药物的研发和审批方面存在重大脱节。FDA肿瘤卓越中心(OCE)的部门主管Harpreet Singh说,“实际上,这种认为每一种新的肿瘤药物在批准时都要对应一种诊断测试,以选择可以安全有效地使用治疗方法的患者的想法并没有发生。”当下的实际情况是什么呢?临床试验开展过程中,各家公司使用本地开发的伴随诊断产品,这些未得到足够的监管。当临床试验的数据被递交到FDA时,FDA才发现没有足够的、合适的患者数据来桥接这些数据。因此,接下来FDA的一个工作目标就是对伴随诊断的监管进一步加强。新药研发企业、CRO企业、生物技术公司都值得特别关注这一新趋势,其背后蕴藏着很大的商机,就如同FDA关于动物试验的决定一样。推荐阅读:世界肝炎日特别活动:动物实验能否被取代?| 药效与安全性-肝病新药研发非临床评价的关键要素世界肝炎日特别活动:动物实验能否被取代?| 药效与安全性-肝病新药研发非临床评价的关键要素08关于现场核查提及FDA进行的现场核查,人们往往立即想到PAI,即Pre-Approval Inspection。除了在批准新药或生物制剂之前进行的这些检查之外,当FDA决定在任何时候需要检查哪些制药工厂时,他们必须权衡几个不同的因素,以优先考虑这些常规的、与质量相关的(cGMP)监督检查。近日,拜登总统最近签署的一项支出法案附带了《食品和药物综合改革法案》(FDORA), FDA现在已经更新了其基于风险的模型,增加了一个关于企业所在国家或地区企业合规历史的风险因素。095000万美元!FDA与两家高校加强合作,建立新的研究中心近日,FDA向北卡罗来纳州两所著名大学,杜克大学和北卡罗来纳大学教堂山分校,拨款5000万美元,用于建立一个监管科学研究中心。这笔资金将于9月1日发放。这笔周期为5年的5000万美元拨款将允许杜克大学和北卡罗来纳大学教堂山分校建立监管科学与创新卓越研究三角中心(CERSI)。该计划还包括与北卡罗来纳州立大学和北卡罗来纳州中央大学的合作。值得一提的是,杜克大学(Duke University)是FDA局长罗伯特·卡利夫(Robert Califf)的母校和前雇主。108200万美元!FDA资助MIT建立mRNA连续化制造平台在联邦政府的帮助下,麻省理工学院(MIT)将致力于设计和安装一个新的连续化mRNA制造平台。根据麻省理工学院报道,该项目得到了FDA生物制品评估和研究中心(CBER)为期三年的8200万美元的支持,旨在通过为其它公司提供一个连续化生产模板来推进mRNA治疗领域,同时促进整个生物制药领域的合作。11FDA检查导致印度工厂自愿关闭,美国从中国进口更多化疗药物(图片来源:齐鲁制药官网)美国正计划从中国进口更多剂量的化疗药物顺铂。美国FDA发言人于7月10日证实,FDA将允许中国齐鲁制药额外再销售10批常见抗癌药顺铂进入美国。负责在美国分销中国产顺铂的仿制药公司Apotex的发言人表示,这10批药将于本周晚些时候从中国运到美国并进行分销。齐鲁的顺铂在今年 5 月份首次获得 FDA 许可进入美国,这些药品未通过正常途径获得FDA批准,在药物标签上存在一些差异。这一点在齐鲁给医疗卫生人员的信(截图如下)中也有体现。(图片来源:FDA官网)根据FDA药品短缺网站的信息,齐鲁先期已经供应了一部分顺铂,还有更多供应批次待定。(图片来源:FDA官网)从今年春季开始,美国的顺铂就一直供不应求。导致短缺的重要一环是FDA在去年11月发现印度Intas药业的一家位于古吉拉特邦的药厂存在质量和数据可靠性问题,这家工厂占美国顺铂供应量的一半。Intas停止了顺铂和一种名为卡铂的类似药物的生产。FDA五月初对Intas的这家工厂再次进行了检查,检查结果依旧揭示了一系列违规行为。6月1日FDA对该工厂发布了进口禁令,但是将涉及短缺的多种抗癌药排除在禁令之外,包括部分顺铂、卡铂、氟尿嘧啶和多西他赛。12武田自愿撤回四价登革热候选疫苗的上市申请美国东部时间2022年11月22日,武田制药宣布美国FDA已接受其在研四价登革热候选疫苗TAK-003的生物制品许可申请(BLA),并授予优先审评资格。在美国,TAK-003正在被评估对4~60岁人群中由任何登革热病毒血清型引起的登革热的预防作用。2023年,FDA要求武田制药提供更多的数据。7月11日,武田制药宣布,基于公司在当前的审查周期内无法满足FDA的要求,决定自愿撤回TAK-003的BLA申请。值得注意的是,2022年8月这款疫苗已在印尼获得全球首批,用于6~45岁人群以避免任何血清型的登革热感染。13“药王”修美乐的一个生物类似药竞争对手被FDA拒绝艾伯维旗下的Humira(修美乐)是近十年来最畅销的药物,在二十年里累计收入达到2000亿美元,2022年,修美乐的收入高达212.37亿美元,再次创下历史新高。然而,“药王”的宝座面临着方方面面的挑战,既有来自K药等新一代创新药的挑战,也有来自专利悬崖带来的仿制药/生物类似药的“蚕食”,艾伯维一直在努力阻挡这些“蚕食”。修美乐的首个类似药,安进生物的Amjevita,已经于2023年1月底在美国上市,另外还有7个生物类似药已经蓄势待发,预计7月份将陆续上市。其中一家是Alvotech。近日,Alvotech收到FDA的完整回复函,其开发的修美乐类似药AVT02的第二次BLA被拒,AVT02是一种仿制修美乐的高浓度生物类似药候选药物。Alvotech称,最新的申请附带了更多的数据和信息。但CRL指出,FDA在3月份对Alvotech位于冰岛雷夫基亚维克的工厂的重新检查中发现了“某些缺陷”。这些缺陷必须在获得批准之前得到解决。Alvotech指出,FDA没有提及其最新申请中的其它问题。14FDA拒绝批准一款帕金森病缓释胶囊在纳斯达克上市的生物制药公司Amneal Pharmaceuticals (NYSE: Amneal) 致力于治疗帕金森病的缓释胶囊IPX203的研发。7月3日,公司表示收到FDA发来的完整回复函(CRL),FDA基于该药物的安全性数据不足,拒绝批准公司用于治疗帕金森病的缓释胶囊IPX203新药申请(NDA),该胶囊含有两种长期用于治疗神经疾病的药物。15FDA拒绝批准一款白血病新药的上市申请瑞典生物技术公司Xspray Pharma近日表示,公司已收到FDA关于其白血病治疗药物Dasynoc上市申请(NDA)的完整回复函。根据PDUFA日期,该药物预计将于今年下半年获批上市。FDA希望该公司向医生和用户提供更多关于Dasynoc剂量的信息,以及FDA正在进行检查的“第三方制造设施”的相关信息。16FDA取消了Curis公司一款白血病新药的临床暂停(图片来源:公司官网)Curis, Inc. (Nasdaq: CRIS)是一家专注于开发emavusertib的生物技术公司,emavusertib是一种口服小分子三靶点(IRAK4, FLT3和CLK)抑制剂,用于治疗血液系统恶性肿瘤。去年,由于一名患者死亡,原因为横纹肌溶解,FDA两次部分暂停了针对白血病患者的1/2期临床试验TakeAim。7月6日,公司宣布FDA已经取消了emavusertib的部分临床暂停。这意味着,Curis可以全力推进该临床试验。此外,emavusertib作为单药治疗的推荐2期剂量(RP2D)已确定为300 mg BID,用于急性髓性白血病(AML)或骨髓增生异常综合征(MDS)患者。17FDA现场核查导致再生元重磅药物阿柏西普8mg制剂被拒6月27日,著名的制药巨擘再生元(纳斯达克:REGN)发布新闻稿,宣布已收到FDA就阿柏西普8mg制剂的上市申请发出的完整回复函(CRL)。公司表示,FDA此次拒绝批准是因为在第三方药物灌装机中发现了生产问题,与疗效、安全性、说明书和原料药生产等方面无关,FDA也没有要求提供额外的临床数据或额外开展试验。(图片来源:公司官网)阿柏西普是一款重磅炸弹级药物,商品名Eylea,用于治疗湿性年龄相关性黄斑变性(wAMD)、糖尿病性黄斑水肿(DME)和糖尿病性视网膜病变(DR) 。根据再生元财报,Eylea在2022年的销售额达到96.47亿美元,同比增长4%,其中美国地区收入62.65亿美元,美国以外市场收入33.83亿美元(拜耳负责,再生元分得13亿美元)。再生元表示,将与FDA和第三方供应商密切合作,尽快将阿柏西普8 mg制剂带给wAMD、DME和DR患者。受此消息的影响,再生元的股价一度下跌近9%,彼时市值786亿美元。18FDA接下来可能批准的重磅药物7月份,FDA还会批准哪些新药呢?根据PDUFA的预期目标日期,预计7月,美国FDA将对4款创新药的批准做出监管决定。(图片来源:药明康德)参考资料:FDA官网Endpoints News、Fierce Biotech等外媒公司官网、官微FDA 专家会改革临近?新研究表明会议数量正在减少(识林)FDA 发布细胞和基因治疗产品生产变更和可比性指南草案(识林)美国首个非处方避孕药获FDA批准上市(医药魔方)首个!一针显著减少出血,血友病基因疗法获FDA批准(药明康德)FDA 再次允许从齐鲁进口更多抗癌药(识林)保护效力长达四年半!武田登革热疫苗在美国申报上市(亿欧)2023年美国市场专利到期的10款畅销药物(求实药社)速递!FDA拒批一款帕金森病新药(药融圈)FDA拒绝批准再生元的阿柏西普8mg制剂(药研界面)FDA拒批再生元阿柏西普8mg上市申请(生物药大时代)7月4款创新药有望获FDA批准(药明康德)FDA 上半年批准的新药汇总(药时代)2023下半年FDA即将批准的10款重磅药物(药时代)FDA正式批准Leqembi上市(药时代)Nature系列综述:21世纪以来FDA批准癌症疗法的趋势(药时代)2023上半年FDA批准肿瘤药物分析(药时代)史上第二贵的基因治疗产品!DMD首款一次性基因疗法ELEVIDYS获FDA加速批准上市(药时代)Review:近20年FDA针对癌症疗法的批准趋势(药时代)其它药时代文章其它公开资料摘要图来源:123rf精彩推荐谁能解决奥希替尼耐药问题?资本寒冬里的好消息!两家新药研发公司在纳斯达克IPO!市场反应积极!价值超70亿美元!辉瑞二度出售潜在“First-in-Class”,为何?世界肝炎日特别活动:动物实验能否被取代?| 药效与安全性-肝病新药研发非临床评价的关键要素不管怎么说,百济还是TIGIT赛道的头号玩家......CM082终于上市了!能治眼底病吗?——CM082故事连载之四雅创医药发表J Med Chem文章:新一代高度差异化FXR激动剂HPG1860的临床前研发点击这里,发现“药时代九万里”!
基因疗法上市批准并购细胞疗法引进/卖出
100 项与 DMD PHARMACEUTICALS 相关的药物交易
登录后查看更多信息
100 项与 DMD PHARMACEUTICALS 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年10月06日管线快照
无数据报导
登录后保持更新
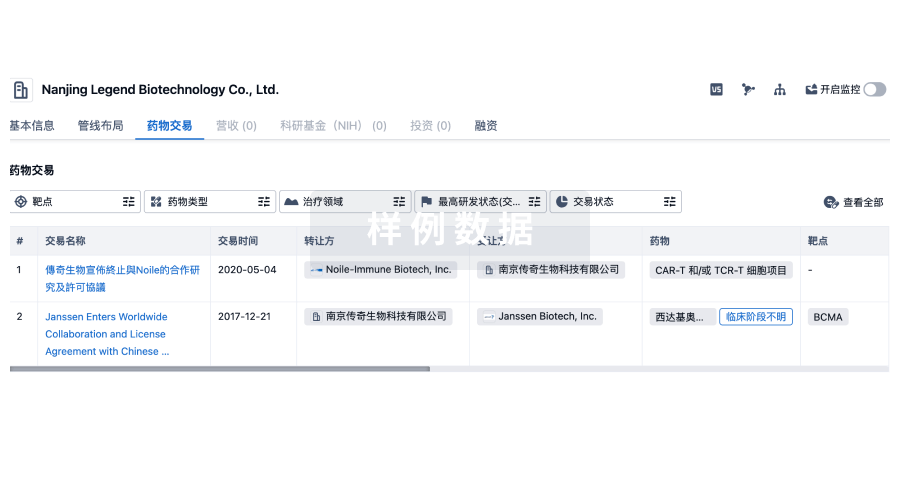
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
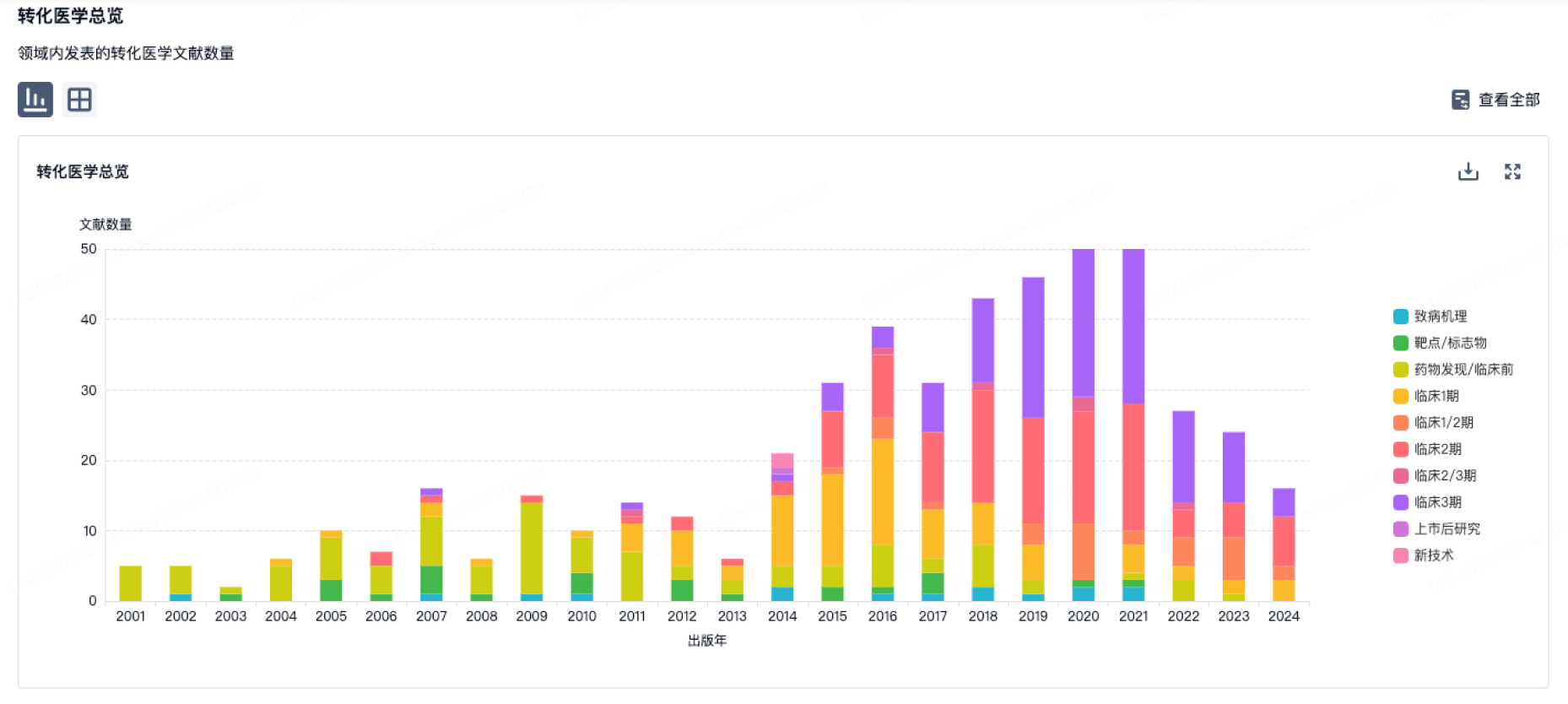
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
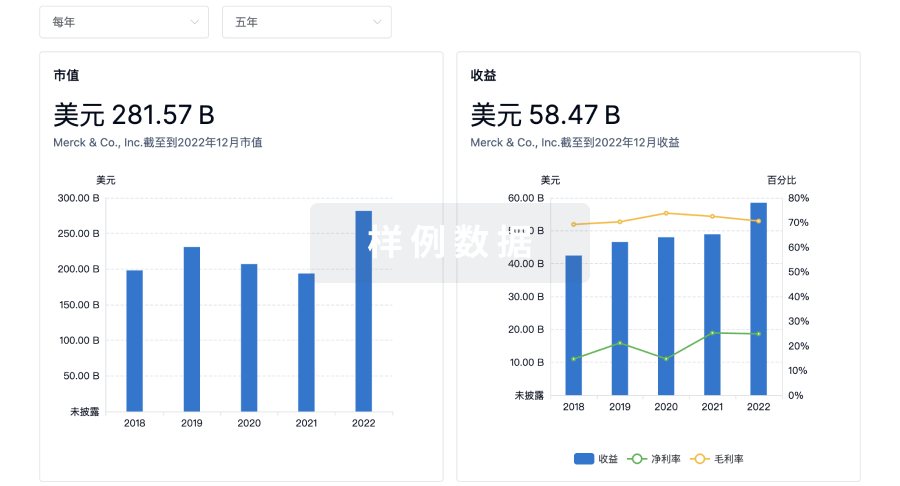
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
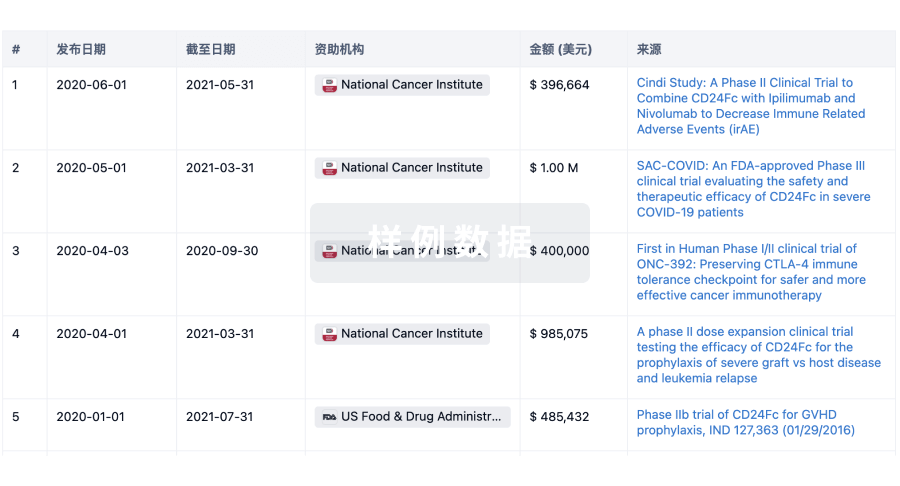
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用