预约演示
更新于:2025-08-21
JY-54
胰岛素类似物
更新于:2025-08-21
概要
基本信息
原研机构 |
在研机构 |
非在研机构- |
权益机构- |
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)临床前 |
特殊审评- |
关联
100 项与 胰岛素类似物 相关的临床结果
登录后查看更多信息
100 项与 胰岛素类似物 相关的转化医学
登录后查看更多信息
100 项与 胰岛素类似物 相关的专利(医药)
登录后查看更多信息
6
项与 胰岛素类似物 相关的新闻(医药)2025-05-14
点击“蓝字”关注我们编者按胰岛素治疗是控制高血糖的重要手段。1型糖尿病(T1DM)患者需依赖胰岛素维持生命;2型糖尿病(T2DM)虽不需要胰岛素来维持生命,但当口服降糖药效果不佳或存在口服药使用禁忌证时,仍需使用胰岛素控制高血糖,减少糖尿病并发症的发生风险[1]。然而,全球范围内,胰岛素在T2DM中使用存在诸多挑战,未能充分发挥其重要作用。近期,加拿大LMC内分泌中心内分泌学家Harpreet Bajaj博士与瑞典瓦斯特曼兰医院初级保健医生Boris Klanger博士做客“胰岛素聚焦(insulin in focus)”系列访谈,两位专家围绕“胰岛素——T2DM治疗中未被充分利用的伙伴”展开对话,分享各自观点。着眼自然病程:T2DM患者为何需要胰岛素治疗?Boris Klanger博士表示,胰岛素是T2DM治疗的一部分。原因在于,随着T2DM病程进展[2],大多数患者最终会依赖胰岛素。T2DM确诊时,通常已丢失50%的β细胞功能。UKPDS研究表明,新诊断T2DM患者即使接受治疗,β细胞功能也会在几年内持续下降[3]。数十年后,T2DM患者会越来越像T1DM患者那样对胰岛素产生依赖。T2DM的进展性也意味着随时间推移,血糖控制目标愈发难以实现,需要调整或强化治疗,最终不可避免使用胰岛素[4,5]。回顾历史:胰岛素经历了怎样的研发创新?Harpreet Bajaj博士指出,β细胞功能障碍随糖尿病病程进行性加重,需要起始胰岛素治疗。胰岛素制剂有多种,起始胰岛素治疗时需根据患者具体情况选择,通常以基础胰岛素为主。回顾历史,胰岛素于1921年由多伦多的Banting和Best发现,该发现于1923年获得诺贝尔奖,授予Banting和MacLeod。基础胰岛素的演变经历了鱼精蛋白锌胰岛素、中性鱼精蛋白Hagedorn(NPH)胰岛素、慢胰岛素(Lente胰岛素),之后长效胰岛素类似物于2000年左右问世,2015年升级至第二代。然而,即使是第二代长效胰岛素类似物,其体内作用时间也维持不到2天。因此,基础胰岛素仍然需要每天注射。令人欣喜的是,2024年起每周一次的胰岛素类似物已在部分国家和地区陆续上市,其半衰期延长,作用时间持续长达一周,极大地帮助患者摆脱每日注射胰岛素的负担。聚焦当下:指南共识推荐基础胰岛素如何使用?Boris Klanger博士分享了《2022美国糖尿病协会(ADA)/欧洲糖尿病研究协会(EASD)T2DM高血糖管理共识》[5]对于基础胰岛素用于T2DM管理的推荐。共识明确指出,当T2DM患者血糖未达标且胰岛素是最佳选择时,不应延迟其起始应用。起始胰岛素治疗时,在现有降糖药物基础上,优先从基础胰岛素开始,并及时调整剂量,以达到个体化空腹血糖目标。Klanger博士指出,在瑞典,由于经济原因,推荐中效胰岛素作为胰岛素的一线治疗选择,只有在患者反复低血糖发作时,才建议转换使用长效胰岛素或长效胰岛素类似物。当然,在其他国家,胰岛素治疗的一线推荐是长效胰岛素和长效胰岛素类似物。剖析现状:临床实践中基础胰岛素治疗存在哪些挑战?Harpreet Bajaj博士讲到,尽管有了这些进展和指南推荐,以及病理生理学的合理性,但T2DM患者起始胰岛素治疗仍存在诸多挑战。有数据显示,欧美国家T2DM患者中,从病程早期到晚期,可能仅约50%患者实现HbA1c达标,尽管他们会从胰岛素治疗优化血糖控制中获益。因此,胰岛素治疗中存在治疗惰性,既有医护人员相关因素,也有患者相关因素。而且,这种治疗惰性在T2DM患者起始胰岛素治疗时尤为明显,会导致血糖控制不佳或无法达最佳状态,进而带来多种并发症。Boris Klanger博士完全同意Bajaj博士的观点。他的临床经验是,T2DM患者现在对胰岛素使用的需求比几年前要晚得多,因为有了更多降糖药物选择,如胰高糖素样肽-1受体激动剂(GLP-1RA)、钠-葡萄糖共转运蛋白2抑制剂(SGLT2i)、二肽基肽酶Ⅳ抑制剂(DPP-4i)等,这也使很多患者未到需要胰岛素治疗的阶段。而且,体重增加及低血糖风险、方案复杂性,也是限制胰岛素使用的障碍。因此,很多医护人员和患者倾向于延迟胰岛素治疗。当前指南也建议,一般来说,在尝试其他方法无效后,胰岛素是最后的选择。然而,有时胰岛素会被遗忘,使用为时已晚。很多医生已失去使用胰岛素的习惯,治疗经验不足,可能导致无法实现以患者为中心的个体化治疗。Klanger博士认为,当T2DM患者HbA1c未达标时,胰岛素应作为一种选择。我们越来越习惯使用其他疗法达到这些目标,这是有风险的。胰岛素延迟治疗可能导致T2DM患者发生不必要的并发症,包括大血管和微血管并发症,以及生活质量下降和死亡率增加。当医疗资源用于治疗并发症而非预防并发症时,还会导致医疗保健系统的成本增加。Harpreet Bajaj博士深以为然。他补充道,T2DM患者未及时起始胰岛素治疗,会使血糖无法达标,导致进行性并发症,并在糖尿病晚期变成永久性并发症,而这是完全可以避免的。总结Boris Klanger博士最后对本次对话进行总结发言,他指出,总之,别忘了使用胰岛素。对于T2DM患者来说,胰岛素仍是一种非常有价值的治疗方法,尤其对于病程较长而其他治疗方案无效者。重要的是,医护人员不要忘记如何使用胰岛素,并尽可能克服医生和患者使用胰岛素的障碍。下期预告下一期“胰岛素聚焦(insulin in focus)”系列访谈中,墨西哥国家医学与营养科学研究院内分泌学家兼高级研究员Roopa Mehta博士与土耳其阿奇巴登大学医学院初级保健医生Pınar Topsever博士将围绕“2型糖尿病基础胰岛素治疗的最新进展”展开对话,敬请期待!参考文献(上下滑动可查看)1. 中华医学会糖尿病学分会. 中国糖尿病防治指南(2024版). 中华糖尿病杂志. 2025; 17(1): 16-139.2. Adapted from: Lorenzo PI, et al. Int J Mol Sci. 2021; 22(8): 4239.3. U.K. Prospective Diabetes Study Group. Diabetes. 1995; 44(11): 1249-1258.4. Leibowitz G, et al. J Diabetes Investig. 2011; 2(2): 82-91.5. Davies MJ, et al. Diabetes Care. 2022; 45(11): 2753-2786.声明:本文仅供医疗卫生专业人士了解最新医药资讯参考使用,不代表本平台观点。该等信息不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议,如果该信息被用于资讯以外的目的,本站及作者不承担相关责任。最新《国际糖尿病》读者专属微信交流群建好了,快快加入吧!扫描左边《国际糖尿病》小助手二维码(微信号:guojitnb),回复“国际糖尿病读者”,ta会尽快拉您入群滴!(来源:《国际糖尿病》编辑部)版权声明版权属《国际糖尿病》所有。欢迎个人转发分享。其他任何媒体、网站未经授权,禁止转载。
细胞疗法
2025-03-24
点击“蓝字”关注我们
引言
备受全球瞩目的心血管领域盛会——第74届美国心脏病学会科学年会(ACC.25)即将于当地时间3月29~31日在美国芝加哥盛大召开。本届会议的重磅研究之一——口服司美格鲁肽片心血管结局研究(CVOT)——SOUL研究的完整研究结果将被揭晓。作为全球首个口服胰高糖素样肽-1受体激动剂(GLP-1RA)的CVOT,SOUL研究结果将可能重塑2型糖尿病(T2DM)合并动脉粥样硬化性心血管疾病(ASCVD)和/或慢性肾脏病(CKD)患者的治疗格局,为心-肾-代谢疾病(CKM)管理提供新的循证证据,敬请关注!
SOUL研究亮点&看点
首个口服GLP-1RA类药物大规模心血管结局研究:扩展GLP-1RA类药物给药途径,验证口服剂型对心血管硬终点的保护效力;
心肾代谢异常广泛覆盖,贴近真实世界高危人群:研究纳入T2DM合并ASCVD和/或CKD患者9650例,样本量巨大,覆盖全球五大洲,亚裔人群占比近1/4(23.4%),较高比例患者已接受降糖、降压、调脂、抗凝等标准治疗,人群具有广泛代表性;
优效性研究设计,复合终点扩展至心肾结局:不同于以往降糖药CVOT多采取非劣效性设计,SOUL研究主要终点——主要心血管不良事件(MACE)复合终点(包括心血管死亡、非致死性心肌梗死、非致死性卒中)采取优效性设计,次要终点扩展至心肾结局复合终点,全面评估药物对心-肾-代谢轴的多靶点保护潜力。
关键结果提前披露,MACE风险降幅达14%:2024年10月SOUL研究初步披露的主要研究结果显示,司美格鲁肽片较安慰剂显著降低T2DM合并ASCVD和/或CKD患者的MACE风险达14%,具有统计学显著性和优效性[1]。
一、 从注射到口服,司美格鲁肽引领心肾代谢管理新纪元
随着T2DM治疗迈入精准化、多靶点干预的新时代,司美格鲁肽作为GLP-1RA领域的革新者,正以“注射+口服”双剂型布局改写疾病管理格局。SUSTAIN 6研究首次证实其注射剂型显著降低T2DM患者MACE风险达26%(HR 0.74;95%CI:0.58~0.95)[2],FLOW研究证实其突破性降低肾脏复合事件风险达24%(HR 0.76;95%CI:0.66~0.88)[3],SOUL研究则将进一步探索其口服剂型对T2DM患者心肾结局的影响。司美格鲁肽不断突破传统降糖药物的治疗边界,在T2DM糖尿病管理正式进入以器官保护为核心的“后血糖时代”,其每次剂型更新和循证突破,都将为全球T2DM管理赋予新动能。
二、 SOUL研究:优效性设计,广泛人群覆盖,直击心肾结局
SOUL(Semaglutide cardiOvascular oUtcomes triaL)是一项国际多中心、随机、双盲、平行分组、安慰剂对照的Ⅲ期CVOT,覆盖全球33个国家和地区444个研究中心。研究于2019年6月至2021年3月纳入9650例T2DM合并ASCVD和/或CKD患者。所有患者在已接受标准治疗的基础上,按照1:1比例随机接受口服司美格鲁肽片14 mg或安慰剂每日一次治疗。研究采用事件驱动的优效性设计(至少1225个事件),持续时间长达3.5~5年[4]。
主要终点为首次发生MACE的时间(由心血管死亡、非致死性心肌梗死、非致死性卒中构成的复合终点);验证性次要终点包括首次发生心肾复合终点(心血管死亡、肾脏相关死亡、eGFR持续降低≥50%、eGFR持续<15 ml/min/1.73m2、启动肾脏透析或肾脏移植在内的慢性肾脏替代治疗)的时间、心血管死亡发生的时间、首次发生重大肢体不良事件(由急性肢体缺血住院治疗、慢性肢体缺血构成的复合终点)的时间(图1)。
图1. SOUL研究设计
根据已公布的SOUL研究基线特征,其具备以下特点[4]:
年龄较大、糖尿病病程较长、体型偏胖、男性比例高,亚裔人群占比可观:平均年龄66.1岁,糖尿病病程15.4年,BMI 31.1 kg/m2,男性占71.1%,亚裔人群占23.4%;
心肾疾病分布具有代表性,体现人群高风险特征:冠状动脉疾病占70.7%,脑血管疾病占21.1%,外周动脉疾病占15.7%,心衰占23%,CKD占42.3%。
标准背景治疗较充分,反映真实临床实践:降糖药物应用比例为二甲双胍75.7%、胰岛素/胰岛素类似物50.5%、磺脲类药物29.1%、钠-葡萄糖共转运蛋白2抑制剂(SGLT2i)26.7%、二肽基肽酶-4抑制剂(DPP-4i)23.0%;主要心血管药物应用比例为血管紧张素转换酶抑制剂/血管紧张素Ⅱ受体拮抗剂/血管紧张素受体脑啡肽酶抑制剂80.0%、β受体阻滞剂63.8%、他汀类药物85.5%、抗血小板药物76.3%、抗凝药物9.4%。
深入分析SOUL研究人群,其与既往其他GLP-1RA的CVOT人群基本一致,同时反映了目前真实世界临床实践注重疾病综合管理的特征,患者降糖、降压、调脂、抗凝手段充分。SOUL研究主要终点采取优效性设计,正是在当下T2DM管理强调CKM全面获益理念下直面问题,重视患者真正获益的体现。即将在ACC.25年会上揭晓的SOUL研究完整结果,将全面展示口服司美格鲁肽片继往开来的心肾保护效力。
三、关注ACC.25研究发布时间,期待口服司美格鲁肽片为T2DM综合管理带来新突破
美国中部时间2025年3月29日下午1:30(北京时间3月30日凌晨2:30),在本届ACC年会的“Late-Breaking Clinical Trials III”专场上,SOUL研究的全球主要研究者(PI)——美国得克萨斯大学Darren K. McGuire教授将揭晓SOUL的完整研究结果。
参考文献:
1.https://www.novonordisk.com/news-and-media/news-and-ir-materials/news-details.html?id=171480.
2. Marso SP, et al. N Engl J Med. 2016; 375(19): 1834-1844.
3. Perkovic V, et al. N Engl J Med. 2024; 391(2): 109-121.
4. McGuire DK, et al. Diabetes Obes Metab. 2023; 25(7): 1932-1941.
声明:本文仅供医疗卫生专业人士了解最新医药资讯参考使用,不代表本平台观点。该等信息不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议,如果该信息被用于资讯以外的目的,本站及作者不承担相关责任。
最新《国际糖尿病》读者专属微信交流群建好了,快快加入吧!扫描左边《国际糖尿病》小助手二维码(微信号:guojitnb),回复“国际糖尿病读者”,ta会尽快拉您入群滴!
(来源:《国际糖尿病》编辑部)
版权声明
版权属《国际糖尿病》所有。欢迎个人转发分享。其他任何媒体、网站未经授权,禁止转载。
临床结果临床终止CSCO会议临床研究
2024-11-27
·医药观澜
▎药明康德内容团队报道
根据九源基因近日公告,该公司将于今日(11月28日)正式在港交所IPO。九源基因成立于1993年,专注于骨科、代谢疾病、肿瘤及血液四大治疗领域。根据招股书介绍,该公司已经建立起多元化的产品组合,包括8款已上市产品,以及超过10款在研产品(包括已递交NDA申请的司美格鲁肽生物类似药JY29-2)。
根据九源基因招股书,该公司已上市产品包括于骨科、肿瘤及血液领域的1款创新药械组合、2款生物制品,以及5款化学药品。其中骨优导是一款含rhBMP-2的创新药械组合产品及骨修复材料。rhBMP-2是一种骨诱导蛋白,可以促进骨和软骨的形成,潜在治疗领域包括骨缺损、骨不连、骨延迟愈合或不愈合的填充修复,以 及脊柱融合、关节融合及矫形植骨修复。
九源基因亦有近18年开发代谢疾病药物的经验,其于2005年开展GLP-1受体激动剂的研究,并凭借多肽药物技术平台开发出利拉鲁肽生物类似药。通过与中美华东的合作,该产品分别于2023年3月及6月在中国获批用于治疗2型糖尿病、肥胖及超重。
九源基因还正在开发另一款GLP-1受体激动剂,即司美格鲁肽生物类似药JY29-2。该产品已经完成3期临床试验,并向中国国家药监局(NMPA)递交治疗2型糖尿病的NDA申请,针对肥胖症及超重适应症也处于3期临床。
根据招股书资料,除了已获批的产品,九源基因正在推进包含10多款产品的在研管线。简要介绍如下:
九源基因在研管线(截图来源:九源基因招股书)
JY29-2:司美格鲁肽生物类似药,包括皮下注射和口服片剂剂型。
JY54:为1类创新药多肽药物胰淀素类似物。该产品可通过抑制食欲、减慢肠道清空,抑制胰高血糖素的分泌,拟开发治疗肥胖症及超重。
JY05:度拉糖肽生物类似药,一款长效GLP-1受体激动剂。
JY23:一款将rhBMP-2与多款不同骨传导生物材料结合而成的下一代骨修复材料。与骨优导相比,JY23在临床前研究中展现出更可控的释放能力及更好的骨传导性能。
JY41:罗姆单抗生物类似药,一款骨硬化蛋白抑制剂,拟用于治疗骨质疏松。
JY-06:一款用于治疗肿瘤化疗后中性粒细胞减少症的长效G-CSF产品。九源基因已于2023年5月在中国递交该产品的上市申请,预期于2024年获得批准。
JY47:为1类创新抗体药物SIRPα特异性单抗,拟用于治疗晚期实体瘤。
JY43:达雷妥尤单抗生物类似药,拟用于治疗多发性骨髓瘤。该公司还在开发皮下注射达雷妥尤单抗生物类似药JY43-2。
九源基因招股书表示,预计本次全球发售所得款项将用于重点治疗领域的在研产品持续研发,现有及接近商业化产品的营销及商业化,此外还将用于寻求战略合作,以丰富产品组合。
参考资料:
[1] 02566-九源基因-新上市. Retrieved Nov 26, 2024, from https://data.eastmoney.com/notices/detail/02566/AN202411261641064551.html
[2]九源基因招股书. From https://www1.hkexnews.hk/listedco/listconews/sehk/2024/1120/2024112000020_c.pdf
本文来自药明康德内容团队,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载授权及其他合作需求,请联系wuxi_media@wuxiapptec.com。
免责声明:药明康德内容团队专注介绍全球生物医药健康研究进展。本文仅作信息交流之目的,文中观点不代表药明康德立场,亦不代表药明康德支持或反对文中观点。本文也不是治疗方案推荐。如需获得治疗方案指导,请前往正规医院就诊。
临床3期生物类似药IPO上市批准临床1期
100 项与 胰岛素类似物 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 肥胖 | 临床前 | 中国 | 2024-03-28 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

生物类似药
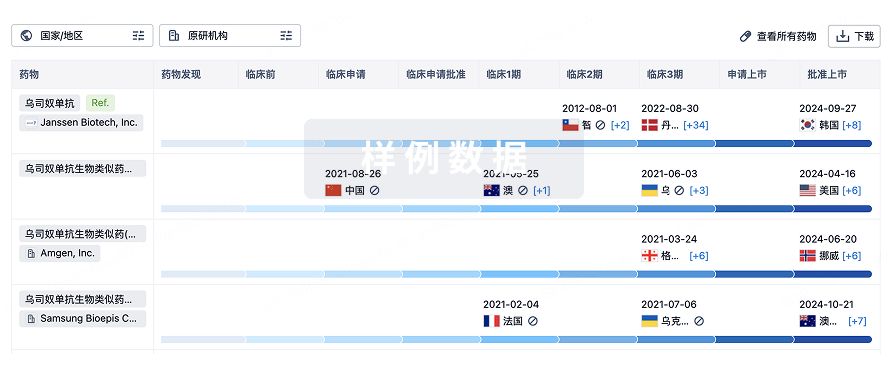
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或

生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用