预约演示
更新于:2025-05-07
Phenindamine Tartrate
酒石酸苯茚胺
更新于:2025-05-07
概要
基本信息
药物类型 小分子化药 |
别名 Phenindamine、Phenindamine tartrate (USAN)、Thephorin + [1] |
作用方式 拮抗剂 |
作用机制 H1 receptor拮抗剂(组胺H1受体拮抗剂) |
治疗领域 |
在研适应症 |
非在研适应症- |
非在研机构- |
权益机构- |
最高研发阶段批准上市 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |
登录后查看时间轴
结构/序列
分子式C19H19N |
InChIKeyISFHAYSTHMVOJR-UHFFFAOYSA-N |
CAS号82-88-2 |
查看全部结构式(2)
研发状态
10 条最早获批的记录, 后查看更多信息
登录
| 适应症 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|
| 超敏反应 | - | - |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
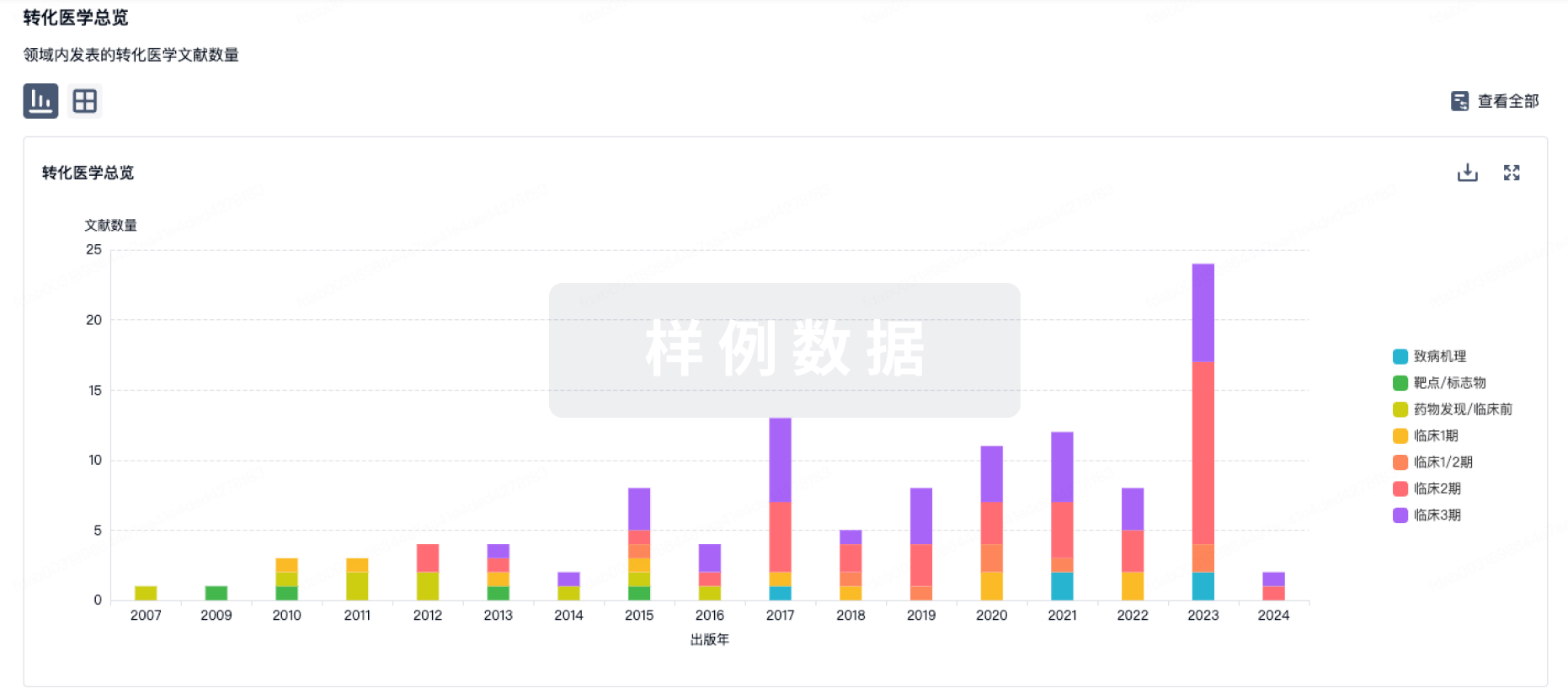
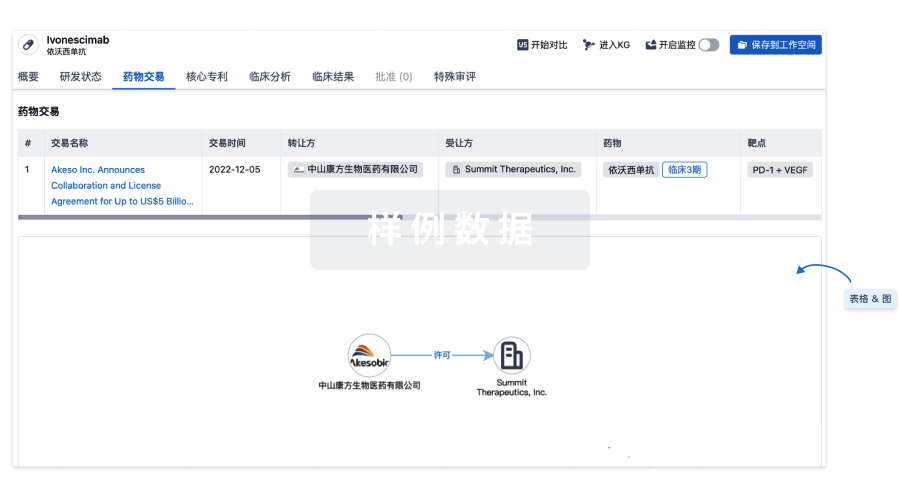
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
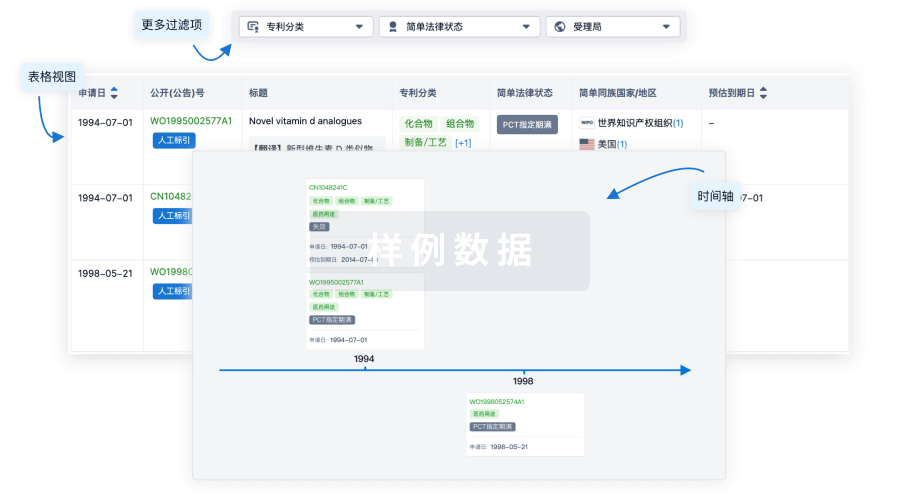
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
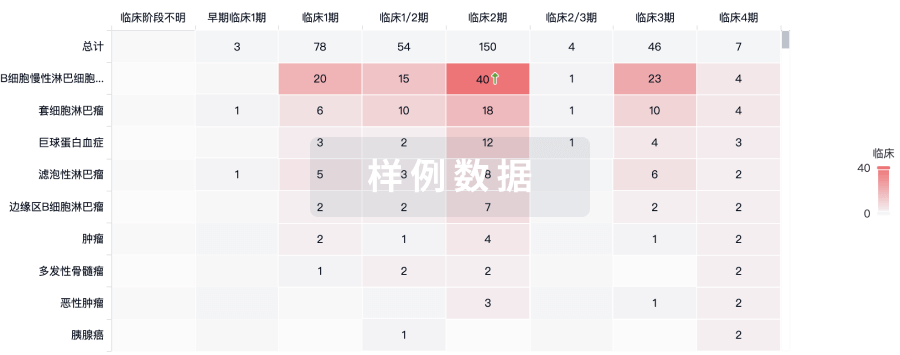
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
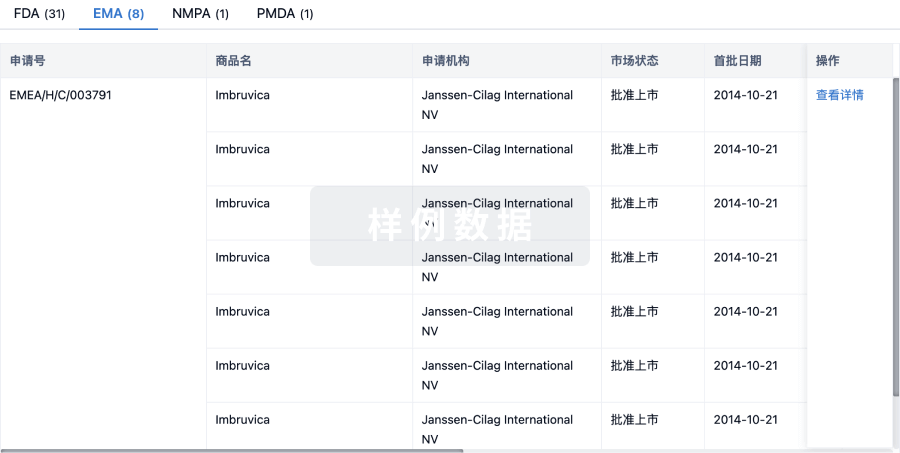
批准
利用最新的监管批准信息加速您的研究。
登录
或
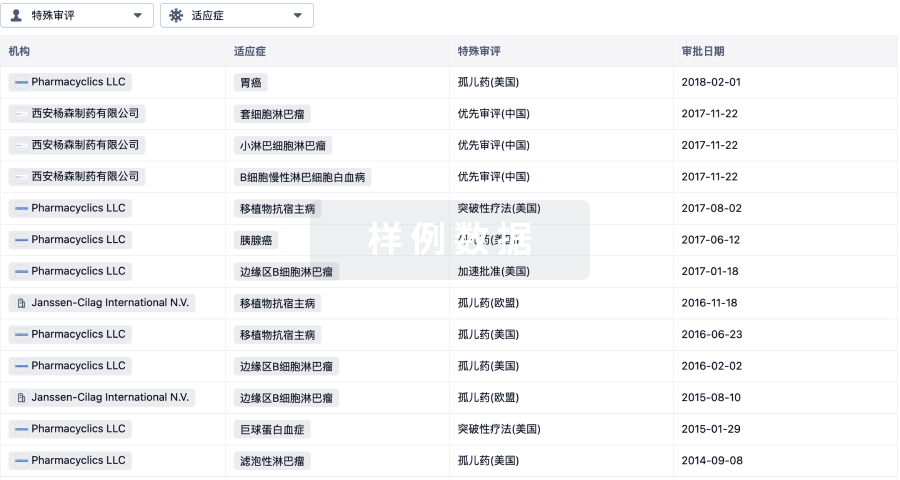
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用