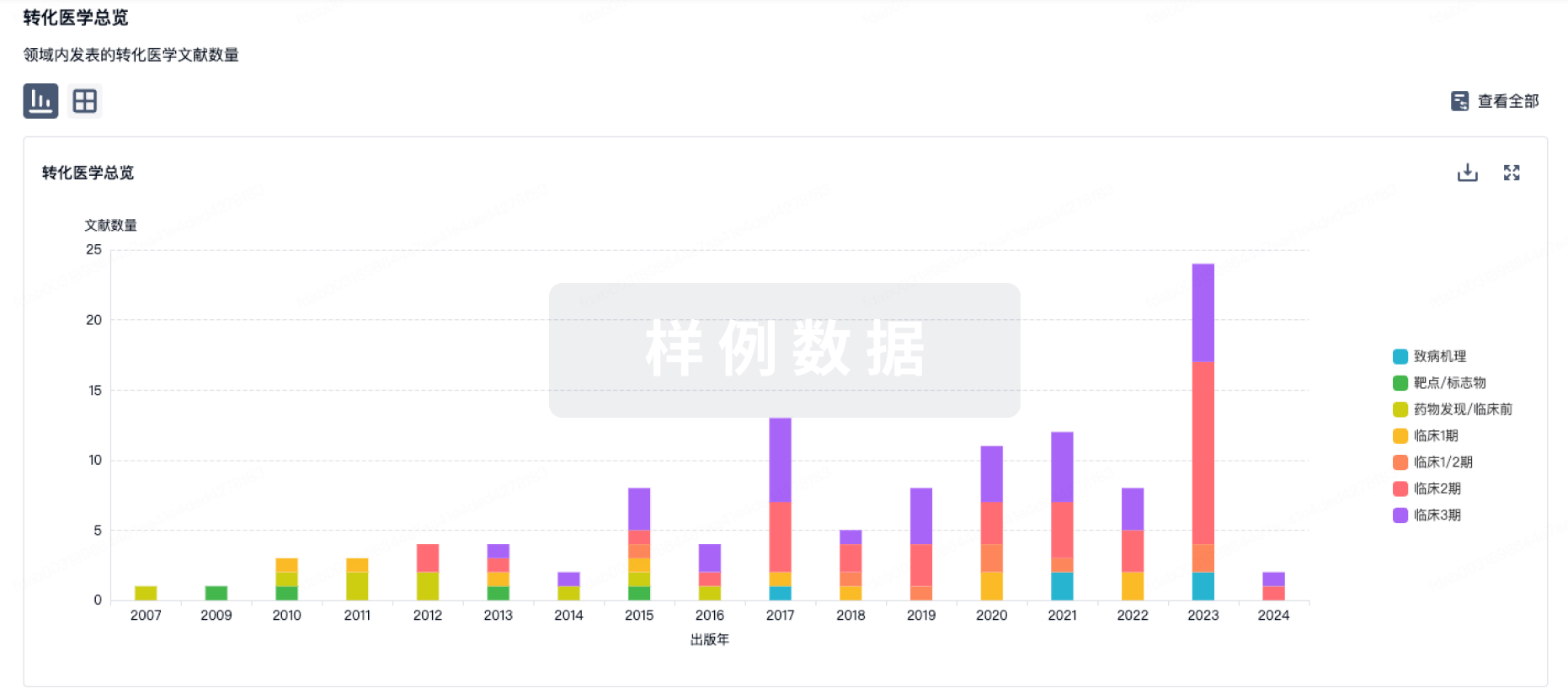
New immunosenescence targets for preventing senescence-associated pathological cardiac hypertrophy (SA-PCH) need to be explored. In the present study, with physiologically aged human and mouse samples, the IL-17A level increased with physiological aging, heart failure (HF), and SA-PCH and was negatively correlated with thymic Bmi-1 expression.
Bmi-1
f/f
LckCre
+
mice and
Bmi-1
f/f
littermates were generated to determine whether Bmi-1 delayed T cell aging by maintaining thymic T cell development to prevent SA-PCH. As a result, Bmi-1 promoted thymic T cell development by upregulating Notch signaling and prevented DN1 T cells from differentiating into γδT17 cells by downregulating γδT17 cell differentiation signaling. Bmi-1 upregulated Notch signaling by inhibiting p53-mediated
Ikzf1
transcription at the −1,863 to −1,849
Ikzf1
promoter region. Bmi-1–RING1B promoted RORγt ubiquitination and degradation by proteasome to inhibit the production of IL-17A in γδT17 cells. Bmi-1 also downregulated
Rorc
transcribed by c-Maf by trimethylating H3K27 at the −1,511 to −1,497
Rorc
promoter region. Subsequently, the number of peripheral γδT17 cells infiltrating the heart tissues was reduced, while alleviating IL-17A-dependent cardiac aging, hypertrophy, dysfunction, senescence-associated secretory phenotype (SASP), and macrophage–myofibroblast transition, ultimately improving SA-PCH. The RORγt inhibitor SR1001 and IL-17A neutralizing antibody ixekizumab prevented thymic RORγt-IL-17A-dependent SA-PCH. Furthermore, RORγt bound to Bmi-1 through ARG237 and to RING1B through GLU235, which could be used as a therapeutic strategy for SA-PCH to construct binding peptides promoting Bmi-1–RING1B binding to RORγt and degrading RORγt for inhibiting γδT17 cell differentiation and IL-17A production. Thus, thymic Bmi-1 prevented IL-17A-dependent SA-PCH by decreasing γδT17 cell numbers.