更新于:2024-09-20
Bevacizumab(Vesselon)
贝伐珠单抗生物类似药(Vesselon, Inc.)
更新于:2024-09-20
概要
基本信息
原研机构 |
在研机构 |
非在研机构- |
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |

序列信息
Sequence Code 41632H

当前序列信息引自: *****
Sequence Code 41637L
当前序列信息引自: *****
关联
100 项与 贝伐珠单抗生物类似药(Vesselon, Inc.) 相关的临床结果
登录后查看更多信息
100 项与 贝伐珠单抗生物类似药(Vesselon, Inc.) 相关的转化医学
登录后查看更多信息
100 项与 贝伐珠单抗生物类似药(Vesselon, Inc.) 相关的专利(医药)
登录后查看更多信息
1
项与 贝伐珠单抗生物类似药(Vesselon, Inc.) 相关的文献(医药)2004-06-07·The Medical letter on drugs and therapeutics4区 · 医学
Bevacizumab (Avastin).
4区 · 医学
Article
5
项与 贝伐珠单抗生物类似药(Vesselon, Inc.) 相关的新闻(医药)2023-02-21
A €444m anti-competitiveness fine given to Novartis and Roche has been revoked by the Paris Court of Appeal, which ruled that the two companies did not abuse their market power to boost sales of their shared eye disease drug Lucentis (ranibizumab).
The Autorité de la Concurrence first levied the fine on the companies in 2020, with the French competition authority charging the pair with illegally preventing the off-label use of an older and cheaper alternative drug to treat wet age-related macular degeneration (AMD).
Wet AMD develops when abnormal blood vessels grow into the macula and leak blood or fluid, leading to scarring and rapid loss of central vision. The condition can develop very suddenly and usually first affects people in their 50s and 60s.
Early detection and treatment of wet AMD may help reduce vision loss and, in some instances, early treatment may recover vision.
Treatment usually consists of regular eye injections, such as with Lucentis, to stop the growth of the abnormal blood vessels. Following diagnosis, patients will usually have a loading dose of three injections, once a month for three months, before being assessed to see if more injections are required.
Despite not being approved for wet AMD, Roche’s Avastin (bevacizumab) had been shown in independent trials to be effective in treating the condition and, as the cancer drug is significantly cheaper that Lucentis, there was a push for its off-label use in this indication.
In response, Novartis and Roche launched legal action to block the off-label use of Avastin and, according to the competition authority, worked to restrict access and spread misleading statements about the risks associated with the drug.
The fine has now been overturned, which was originally €385.1m against Novartis and €59.7m against Roche and its subsidiary Genentech, with the appeals court concluding that the companies had been ‘measured in tone’ in their comments about Avastin’s use in AMD.
The court also ruled that Roche’s refusal to supply samples of Avastin for a comparator clinical trial could not have had an anti-competitive effect.
Novartis and Roche have since confirmed the decision, with Novartis reasserting its opposition to the use of off-label products in an unlicensed indication.

专利侵权优先审批
2023-02-18
1.前言首个用于治疗癌症的单克隆抗体-利妥昔,在经过长期的工程化和优化后于1997年批准上市。然而,全长的抗体仍然面临着肿瘤渗透率低,体内循环的长时间滞留,非肿瘤组织的非特异性结合和高昂的生产成本等方面的局限。自从2018-2019年,第一个双特异性抗体贝林妥和纳米抗体卡拉西单抗的相继问世,抗体片段工程在癌症治疗方面表现出了前所未有的优势。现如今,已存在多种抗体衍生的分子构型,例如scFv, Fab,微抗体,双抗体,三抗体,纳米抗体等,这些分子构型提供了一系列优于单抗的优势:可供选择的注射方式,能够进入到单抗分子难以到达的部位去结合抗原,能够穿过血脑屏障,减少由Fc段带来的副作用,在扩大生产方面成本低,稳定性强,工程化和修饰的灵活性高,免疫原性较低。图1 抗体片段工程总结图表2. 抗体片段构型2.1 scFv和纳米抗体scFv是由一条重链可变区与一条轻链可变区相连而成的抗体片段。最普遍的连接肽链是由(G4S)3/(SG4)3构成,这种连接肽链对于调节两个结构域来说足够的灵活。除了G和S,其他氨基酸例如G和A也被引入到连接链中,用于提高分子的灵活性。scFv中引入更长的G4S连接肽链被证实具有更强的结合能力。然而,这样简单的构型由于缺少Fc区域,所以也有一些缺点:scFv并不像全长抗体那样稳定,而且容易聚集和错误折叠。因此,这些抗体片段需要大量的修饰和工程化以满足体内研究的质量要求。纳米抗体,也称作VHH,是由骆驼衍生出来的。纳米抗体是最小的抗体(12-15kDa),只有半个scFv(25kDa)大,并且只由重链可变区组成。纳米抗体与抗原的结合主要是通过CDR3,但是它们的CDR3与抗原结合能够形成一个凹表位。这个特征就使得纳米抗体能够识别常规抗体无法识别的,存在于一些缝隙中的或隐藏起来的一些神秘的表位。另一方面,与抗原相互作用的较短区域增加了分离高亲和力纳米抗体的困难。不过在极端条件下,诸如高温高压、低pH值和低蛋白酶浓度,纳米抗体依旧稳定,这也降低了生产成本。除此之外,延伸CDR3区,将VH中FR2区的保守疏水氨基酸替换成亲水氨基酸以及引入额外的二硫键,这些方法可以使得纳米抗体形成稳定的构象和较少的聚集。2.2 scFV和纳米抗体工程纳米抗体和scFv相较于全长抗体表现出了很多优势。它们由于自身更小的分子量,被证明有较强的穿越内皮和上皮屏障的灌注速率和在肿瘤基质中更快的扩散速度。不过,这么小的scFv纳米抗体仍然有其缺点。例如,纳米抗体15kD的分子量低于肾的过滤限制,使得它们在体内的半衰期极短。这么短的半衰期就限制了纳米抗体的治疗效率和潜力,尤其是在长期的慢性疾病的治疗方面。因此,许多改善scFv和纳米抗体半衰期的技术方案发展起来了,这其中包括在它们的C末端连接一个人源Fc区域,在可变区连接一个抗白蛋白抗体,聚乙二醇化,偶联人血清蛋白,或者是目前的,多聚谷氨酸和PAS化修饰技术。偶联HSA,延长了纳米抗体5-10倍的半衰期,代价是分子量变的更大,达到了50kDa。一些更多的设计也利用了小分子量的优势,将纳米抗体与scFv或者Fab进行组装,这样的构型能够通过参与新生儿FcRn介导的IgG循环通路来延长抗体的半衰期。尽管短的半衰期和缺少Fc区造成了低生物利用率和免疫应答,但是这些特征也使得它们带有更少的副作用的特点。除此之外,较短的半衰期也使得它们成为低背景药物成像的理想分子。2.3 多价抗体片段scFv和单域抗体,能够进一步的工程化为多价抗体片段例如,单抗体,双抗体,三抗体,微抗体以及更多的复杂的构型。双抗体的形成是通过非共价或者共价键将两个带有不多于11个氨基酸肽链的scFv相连,而三抗体和四抗体是在连接肽链含有不多于3个氨基酸。这些构型被证明比单价片段具有更高的特异性和亲和力。更多的是,两个scFv能够通过CH3区连接形成微抗体,这种构型虽然仍旧相对小,但是比其他共价构型更稳定,这使得它成为成像试剂和药物注射的理想分子。例如,放射标记的靶向CD8的微抗体在CD8阳性T细胞的组织检测方面展示出了很好的耐受性和敏感性;因此,它被用于免疫治疗后,监控CD8阳性T细胞的应答。图2 四大抗体类型3.单价构型单价构型的抗体片段的重点在拮抗效应上面,这是因为Fc段的缺乏。纳米抗体和scFv不能够引起免疫应答,包括ADCC和ADCP或者CDC。90年代初期,纳米抗体和scFv在抗癌细胞表面蛋白如cetuximab, panitumumab, trastuzumab, rituximab 和obinutuzumab已有所发展。现今,已有大约700中单克隆抗体药物应用在临床上和临床前研究,其中有106中的单克隆抗体药物已被FDA批准用于癌症的治疗,这对研究可选择的抗体构型目标提供了一项概念的支撑。3.1 EGFR家族EGFR被认为是治疗癌症的主要抗原,因为它在不同的上皮肿瘤细胞中过表达,例如乳腺癌,非小细胞肺癌(NSCLC)和结肠直肠癌(CRC),并且伴随着不良预后。结合EGFR的配体能够引起下游信号通路的激活,包括磷脂酰肌醇三激酶(PI3K), v-akt小鼠胸腺瘤病毒癌基因同源物1或蛋白激酶B (Akt), 促分裂原活化蛋白激酶(MAPK)和 信号转导和转录激活(STAT),这些通路都能够促使肿瘤细胞的增殖,迁移和存活。US FDA批准了4种抗EGFR的单克隆抗体(cetuximab, panitumumab, necitumumaband,amivantamab)用于治疗CRC和NSCLC,这为抗EGFR的scFv和纳米抗体的研究道路奠定了基石。Her2与EGFR有类似的下游信号通路,并且Her2与EGFR密切相关,其主要在乳腺癌和卵巢癌中过表达。它的高表达水平使得其成为治疗乳腺癌的理想靶点。事实上,抗Her2的trastuzumab是最有争议的单克隆抗体药物之一。它能够将患有转移性乳腺癌患者的生存期从20.3个月提高到25.1个月,并且能够将三年内复发的风险降低到10%。尽管抗EGFR/Her2的全长抗体药物取得了成功,但是抗体片段工程的发展空间仍然很大。Fab臂交换的双特异性抗体amivantamab最近获得了US FDA的批准,用于同时靶向EGFR和cMET来治疗NCSCLC。抗Her2的纳米抗体和scFv也被研究来提高抗Her2治疗的肿瘤渗透率。由于缺乏FC段,抗体片段不能执行ADCC或ADCP效应。用于抗肿瘤毒性的抗体片段偶联药物的方法被开发了出来。CAM-H2是131碘偶联的抗Her2的纳米抗体,近期被用于临床I、II期实验,用来治疗发展和转移性的Her2相关的恶性肿瘤。纳米抗体的小分子量,能够使其穿越血脑屏障到达转移的Her2阳性恶性肿瘤细胞,这是传统抗体所无法做到的。Her3在83%的胃肠肿瘤细胞中和20%的乳腺癌,卵巢癌和膀胱癌细胞中过表达。Her3能通过负反馈机制来抵抗Her2的抑制作用,从而维持RAS-Akt信号通路。除此之外,Her3还可能形成异二聚体来促进肿瘤的增殖,这使它成为靶向癌症治疗的一个关键的靶点。Lumretuzumab是开发出来的用于靶向Her3并抑制其磷酸化和配体结合的单克隆抗体。在I期临床实验中,用Lumretuzumab治疗的单一疗法和其他抗Her3的单克隆抗体治疗方案只有21%无ADCC效应的控制率。单域抗体随后被研究通过独特的机制来靶向抗增殖效应。尽管抗Her2和Her3的抗体片段不能执行ADCC或ADCP效应,但是它们能够在靶向Her2和Her3多种表位的多特异性抗体复合物对抗肿瘤的单一药物治疗的这场战役中起到基石的作用。3.2 VEGF和HGF(血管内皮生长因子和肝细胞生长因子)VEGF和VEGF受体在血管生成和血管新生中过表达,这促进了心血管在不同癌细胞中的发展,包括结肠癌,乳腺癌和肺癌。用化疗的方法抑制VEGF和VEGFR显示能够延长此类癌症患者的生存期。2004年,US FDA批准了bevacizumab (Avastin)用于通过中和VEGF来治疗转移性的CRC。Avastin能够阻断VEGF和VEGFR的互作,因此能够促进化疗药物通过血管输送到肿瘤部位。现在,几乎很少有抗VEGF或VEGFR2的纳米抗体被开发出来用于抑制肿瘤的血管生成,并且抗VEGF的纳米抗体会抑制人脐静脉内皮细胞增殖和体外管腔形成。然而,现今没有任何抗VEGF/VEGFR的纳米抗体有体内抗肿瘤效应的证据,这很可能是由于纳米抗体在血液中的半衰期较短的原因。一些研究试图将IgG1的Fc段融入抗VEGFR的纳米抗体中,但是体内效应尚不明确,尽管Fc介导的功能在体外已研究透彻,未来关于抗VEGF/VEGFR的纳米抗体的人源化和亲和力成熟将有可能有助于体内功能性单价纳米抗体的开发。HGF是血纤维蛋白溶酶原类蛋白,能够与cMET受体结合。这导致了酪氨酸激酶的激活,该激酶能够促使肿瘤的发展和转移。最近,五种用于治疗CRC,肾细胞癌,神经胶质瘤, 胃癌和食管癌的单克隆抗体已经进入了临床试验的I-III期。不幸运的是,onartuzumab (anti-cMET) 和rilotumumab (anti-HGF)在治疗高表达cMET的患者的临床III期实验中失败了。尽管cMET在可能不是治疗患者的最佳生物靶点这方面颇有争议,但是cMET和HGF或许存在着不依赖激酶的肿瘤发生通路,这种可能使得抗HGF/cMET单克隆抗体和抗HGF纳米抗体的作用失效。为了证实这一观点,一种抗cMET胞外域的纳米抗体被开发了出来,并且该抗体被证实比起正常组织,它对肿瘤组织有着更高的渗透率,并能够延迟肿瘤的生长。传统治疗癌症的抗体的成功对于研究抗体片段构型是一把双刃剑。新兴的抗体构型的弊端很难忽视:缺少功能和药用潜力的依据,安全问题和半衰期短。但抗体片段仍然被证实是相较于传统抗体拥有者前所未有的优势,以传统抗体作为基石发展起来的多特异性抗体分子和CAR-T/CAR-NK的细胞治疗被赋予了更灵活的分子特征。4. 多特异性构型多特异性抗体分子构型是由两个以上的抗体可变区构成,或者是单独的VH或者是VH+VL。通过与不同抗原依次地或者同时地结合,多特异性抗体构型能够通过不同的机制获得新型的功能。4.1 -4.3 BiTE抗体及其药效与特异性修饰BiTE募集T细胞靶向肿瘤细胞,导致T细胞在CD3与T细胞受体复合物(TCR)的结合作用下被激活。T细胞的激活不受主要组织相容性复合体(MHC)的限制,导致不依赖TCR特异性的靶细胞被破坏。如今,超过15种抗实体瘤的BiTE应用到了临床研究,这些研究的主要靶点是一些已经研究的比较全面的抗原,如EGFR,HER2,PSMA和EpCAM。(见下图)单抗治疗实体瘤比治疗血液肿瘤更为复杂,这是由于单抗在实体瘤中的渗透率很低。BiTE是由scFv或者纳米抗体组成;因此,与全长的IgG相比,小分子量的它们有更高的实体瘤渗透率。例如,抗CD3/EGFR的双特异性纳米抗体特异性地结合到EGFR过表达的肿瘤细胞上,因此,通过激活T细胞和阻断EGFR信号通路,介导了细胞的裂解。由于BiTE抗体片段的分子特征,在减少其毒性的基础上,开发它的特异性,药用潜力,亲和力方面仍有很大的发展空间。为了预防副作用或脱靶效应,新一代的由抗体片段组成的Fc失活或缺失的BiTE正经历着临床研究实验。USFDA于2014年批准了blinatumomab (CD3+CD19)关于急性淋巴细胞白血病(ALL)的治疗;它是一种能识别CD3和CD19的,通过连接肽链将两个scFv连接在一起的双特异性抗体。每日使用低至0.005mg/m2的剂量,就能够消除非霍奇金氏淋巴瘤患者体内的CD19阳性细胞。在临床II期实验中,证实了它对于外周血肿瘤细胞的持续清除作用。所有患者均表现出大于6个月的持续缓解,不良反应如淋巴细胞减少、白细胞减少、细胞因子释放和寒战均可控制。对自身免疫疾病以及细胞因子风暴综合征患者的治疗方案十分匮乏,这也鼓励了研究者未来对相关BiTE方面的展开进一步研究。4.4 双/三特异性杀伤衔接器为了提高特异性和活力,上述构型可以被工程化为抗CD3的同时,带有至少两个特异性抗原结合位点(2+1, 3+1, 4+1)。例如,串联的双抗体CD3+CD19 AFM11(2+2),CD3+CD123(2+2)是目前在开发的,并且由四价的抗CD3和CD19/CD123片段组成。该四价抗体能够同时靶向T细胞,B细胞和骨髓细胞。这种四价分子构型的一个吸引人的特点是能够不依赖T细胞而获得同样的效果:靶标细胞比例。多价分子构型能够减少临床应用中的有效剂量,因此可以提高多特异性抗体治疗的安全性。尽管在体内,四价分子比而二价分子具有超过10倍的潜力,但是临床试验数据表明这种构型就药效而言缺乏足够的优势。三价抗体,例如二价结合CD20,单价结合CD3(2+1)的RG6026,在临床I期实验中,总体获得了42%的完全缓解率,但是有8%的患者经历了不少于3级的细胞因子风暴综合征。scFv和纳米抗体的结合是通过抗CD3的scFv和抗EGFR、EpCAM的纳米抗体搭配而成。这个三特异性T细胞衔接器在体外诱导依赖于T细胞的EGFR-EpCAM双阳细胞的杀伤。设计这些分子构型是期待能够拥有比双价构型分子更高的肿瘤抗原特异性,T细胞裂解能力几乎无副作用。尽管BiTE在淋巴瘤治疗方面取得了成功,但是就癌症免疫治疗而言,特异性和潜力的提升仍有进步的空间。除了BiTE,双特异性和三特异性NK细胞衔接器也被开发出来用于激活NK细胞上的受体和肿瘤相关抗原。NK细胞比T细胞更安全,因为NK细胞注射不会引起移植物抗宿主的疾病。NK细胞表达的CD16是诱导ADCC的基础,该ADCC是通过与肿瘤结合的抗体的FC段引发的。因此,双特异性scFv既结合CD16还结合癌相关抗原,包括CD19,CD20,CD33,CD30和B细胞成熟抗原(BCMA)等。双/三特异性杀伤衔接器促进NK细胞脱粒,这导致不依赖抗体Fc段的癌细胞的裂解加强。AFM13是一个串联的抗CD16A和CD30的四价双特异性抗体,其针对CD30的IC50为35.8nmol/L。I期的临床试验中,高剂量的AFM13在霍奇金淋巴瘤的患者中耐受良好,有高达77%-88%的效应率。AFM13作为单一治疗CD30阳性T淋巴细胞的药物,其药效正在经历II期临床试验(NCT04101331)。为了优化特异性,基于抗CD16A的双特异性抗体也被开发出来,用于促使NK细胞和巨噬细胞通过非特异结合其他的Fc段受体来靶向癌细胞。双特异性NK细胞衔接器也被工程化开发用于合并其他分子,形成具有更强特异性的三特异性NK细胞衔接器和/或增强NK细胞的活力和存活。4.5 其他双特异性抗体参与多种肿瘤细胞信号通路的双特异性scFv也被开发出来,用于通过靶向相同癌细胞上的多种抗原来减少癌症对于单一抗体药物的抗药性。例如,EGFR和Her2的突变被发现存在于15%-30%的NSCLC样品里和1.6%的Her2阳性乳腺癌细胞里。EGFR,Her2和Her3共享相类似的下游信号通路,抗EGFR治疗诱导Her2的过表达,这使得肿瘤对于抗EGFR治疗产生抗性。尽管抗Her2和抗EGFR的单克隆抗体在治疗Her2/EGFR阳性患者方面取得了临床上的成绩,但是只有10%-25%的Her2阳性患者对于trastuzumab的治疗有效果。为了获得高的应答反应率和低的肿瘤耐药性,针对单一治疗没有应答的患者的多特异性抗体的治疗正在被研究。癌症治疗的另一个主要的难关就是肿瘤微环境的免疫抑制。一些方法已经建立用于对抗由于用抗VEGF和Ang2的双特异性纳米抗体联合抗PD-L治疗NSCLC的免疫抑制和肿瘤血管发生。在大多数最近的Ib期临床实验中,12位患者中有两位表现出了部分副作用可控的反应。这些双特异性抗体构型证明了潜在的细胞毒性和抗体浓度的降低。然而,它们对抗低抗原表达的癌症的抗肿瘤效应仍然十分贫瘠。这或许是由于每个抗体的狭窄的表位中和了双特异性抗体的结合能力。4.6 微抗体工程不像全长的免疫球蛋白那样,抗体片段并非必要包含有CH1,CH2或CH3结构域,因为恒定区会增加分子的大小并减小抗体片段的渗入。因此,单独的CH2或CH3结构与被用来替换全长的CH1,CH2或CH3来连接单价抗体片段,以保证稳定性的前提下来减小分子的大小。一个有效的增强单CH2或CH3域的策略就是引入额外的域间或域内的二硫键。二硫键对于免疫球蛋白的热稳定性是最重要的共价键。在CH2中引入R292C/N297G/V302C突变引起额外的域内二硫键,协助提升抗体分子的稳定性,并在体内循环中的降低其清除的速率。CH3域也被工程化,在C末端引入P445G, G446E和K44C用于添加域内和域间的二硫键。糖基化修饰也被应用到CH2结构域中来改造结合FcγR和配体的FC融合抗体片段。N297突变的引入,能够有效的使抗体糖基化,这将应用于由于对FcγR和配体结合降低引起的自身免疫性疾病的治疗。CH2结构域的糖型工程对于Fc融合抗体片段的临床治疗是重要的,因为普遍的细胞系例如CHO,将会在CH2结构域中引入免疫原性的糖基化。Lonza和Roche开发了基因改造的CHO细胞来生产没有免疫原性的并且同源基因糖基化CH2的细胞系。糖基化工程的抗CD20(obinutuzumab [GA101]),证明了对FcγRIII的高亲和力会引起效应功能的提升,这已被批准用于治疗慢性淋巴细胞白血病。4.7 抗体片段多价化scFv和纳米抗体多聚化近年来十分盛行,并且提升了它们的活力。三特异性或四特异性的scFv或纳米抗体已经设计成通过抗相同抗原结合抗体片段。理论上,共价形式能够产生靶向同一抗原多种表位的抗体构型。抗DR5的多价纳米抗体(DR5Nb1 by Ablynx)证明了强大的肿瘤杀伤潜力,其血液半衰期有5-9h,而全长的抗体则大于1周。这减少了其在体内的药效。既它的成功之后,四价的抗GD2scFv片段被开发并PEG化来增加其稳定性和半衰期。PEG化的GD2特异性四价scFv片段比全长的单克隆抗体(dinutuximab)取得了更好的肿瘤滞留和低脱靶效应。仍然,四价scFv的肿瘤细胞毒性和肿瘤生长抑制能力仍需进一步优化。更多复杂的设计,例如脂质体药物包裹的三特异性抗体也已经被证实有优于双特异性抗体的体外活性。被中和的亲和活性和稳定性在多价构型中十分常见。未来对于结合高通量筛选的亲和力和稳定性的优化和研究和深度学习对于多价抗体构型来说至关重要。图3 网络系统高通量筛选双特异性抗体5. Car-T/NK细胞中的scFv和纳米抗体CAR是一种融合蛋白,由细胞间信号结构域,跨膜铰链域和抗原结合域组成。CAR工程化的T/NK细胞在治疗恶性血液疾病上取得了令人鼓舞的成绩。5.1 Car-T/NK细胞中的scFv和纳米抗体CAR-T细胞在特定的癌症类型上取得了令人兴奋的临床成果,例如B细胞白血病和多种骨髓瘤。抗原结合域传统上被设计成scFV或者纳米抗体,来识别癌细胞表面的抗原或者可溶性配体,以诱导不依赖MHC的T细胞的激活。TCR模拟CAR也已经被用来结合细胞内的肿瘤相关抗原,导致依赖MHC的T细胞激活。美国FDA已经批准了五种CAR-T的治疗:Abecma, Breyanzi,Kymriah, Tecartus and Yescarta, 其中四种都是用来治疗淋巴细胞白血病的。Kymriah, Breyanzi和Yescarta利用鼠源抗CD19的FMC63提高患者体内潜在的抗CAR免疫应答的关联。除了治疗血液疾病外,CAR-T治疗实体瘤的应用也被开发了出来。BioNTech Cell 和 Gene TherapiesGmbH开发了一种联合治疗方案,该方案利用抗claudin6的CAR-T细胞通过脂质体包被的mRNA复合物进行电转,来治疗实体瘤,包括卵巢癌,睾丸癌,子宫癌,肺癌和胃癌。这种脂质体包裹mRNA编码Claudine6的复合物被引入树突细胞以促使注射CART细胞的存活和扩大,这将促使体内肿瘤退化。这种抗实体瘤的治疗方案的药效和安全性正在临床I、II期实验中被评估(NCT04503278)。CAR-NK治疗在近些年也获得了更多的关注,因为它们与CART细胞相比,表现出许多优势。NK细胞机体自身固有的淋巴细胞,并没有抗原特异性受体,例如TCR;因此,它们能够杀伤癌细胞或者以不依赖于CAR的固有机制的方式感染细胞。这也使得CAR-NK治疗更安全,因为它不会引起移植物抗宿主并发症。然而,全球只有20个CAR-NK临床试验在进行。clinicaltrials.gov列出了500多个CAR-T临床实验,可能是良好的操作标准和病毒转导成功率欠佳,导致原始NK细胞的体外扩增速率较低。为了阐明这一问题,抗HER2 CAR的NK-92细胞系在临床试验中被评估(NCT03383978)。另一项研究利用脐带血中的原代NK细胞来进行抗CD19的合并,导致无副作用的73%的应答率。CAR工程的有效研究在最近5年发展巨大,同时伴有创新性的scFv和纳米抗体构型合并在CART的治疗中,进入了早期的临床研究。抗原结合结构域(scFv和纳米抗体)是CAR最重要的成分,决定了CART细胞的抗原结合的亲和力和特异性。抗神经节苷酯GD2的scFV中的单个氨基酸的突变完全消除了体内抗GD2 CART细胞治疗的副作用。拥有理想的亲和力和特异性的靶向抗原的CART细胞的开发已经完成,因为CART细胞需要相当大的亲和力来识别肿瘤抗原。这反过来可以诱导T细胞裂解靶细胞;高亲和力也能够导致其结合到健康的组织上。更进一步,高亲和力的CAR-T细胞被证实有较低的体内持续时间,这是因为过度的激活介导了T细胞死亡。尽管抗体融合能够改善这种情况,但是其他CART试剂产生的瘤外毒性限制了它们的临床使用。5.2 应用于降低CART瘤外毒性的scFv/纳米抗体工程与双特异性抗体片段工程相似,亲代单克隆抗体的成功并不预示着CAR的功能。Trastuzumab的可变区被用到了抗Her2 CART细胞中,但因正常组织也表达HER2,仍存在瘤外毒性。一些策略,例如亲和力调优,利用生成CAR的肿瘤和非肿瘤组织上抗原表达的不同的差异进行开发,使其更亲和于肿瘤组织。为了优化CART细胞的安全性和治疗窗口,低亲和力的scFV被用作靶向那些肿瘤高表达但是组织低表达的抗原。这个策略产生了抗CD19和EGFR CART细胞的低亲和力scFV,证明了由于抗原诱导的CART细胞的扩增和体内高特异性抗肿瘤效应的优势导致的更长的持久性。除了亲和力,CAR上的抗体片段构型也影响CART治疗潜力和药效。scFv上可供选择的构型也被研究用来减少基于scFv CAR的免疫原性,聚集和错误折叠。正如先前讨论的,纳米抗体表现出较低的免疫原性,小的分子量,和在极端环境下的显著的稳定性,这使得它们成为构建CAR的理想候选者。随着2019年FDA批准了纳米抗体的单一治疗,更多基于纳米抗体CART治疗的可行性投入到了研究。抗CD19+CD20 CART细胞的双特异性纳米抗体已经形成,并且具有较高的特异性和较强的增值性,在体外杀伤B细胞系。尽管基于scFV的CART治疗血液肿瘤的成功,实体瘤的治疗仍然具有挑战性。其中一个主要的阻碍就是抗原与CAR的结合。另一方面,基于纳米抗体的CART细胞,包括抗CD105,EIIIB和BCMA,能够进入隐藏的表位,这是由于CDR3较长,体内的肿瘤大小较小。纳米抗体一个吸引人的特征在于能够靶标大于一个抗原表位。抗BCMA的CART治疗中,LCARB38M利用能够识别两个抗原表位的纳米抗体推进特异性治疗并诱发了较少的细胞毒性。尽管直接比较临床研究结果比较困难,但是LCARB38M证实了有高达88%的应答率。更进一步的是,LCARB38M在临床上的用药剂量比scfv治疗的剂量要低的多。这些令人鼓舞的早期临床研究给其他基于纳米抗体的CART治疗实体瘤带来了希望。与抗体治疗相比,CART治疗面临着更多的困难,在靶标抗原表达很低的时候。这一问题在CART细胞用于治疗实体瘤的时候更为严峻,因为它们不能接近抗原以及受免疫抑制微环境的影响。5.3 用于提高特异性的CATR治疗中多价scFv的应用单价CART治疗在治疗血液恶性肿瘤上已经取得了成功,但是仍然面临着一些限制,例如六个月后的高复发率,这很可能是由于CART细胞的低持续性或者是通过不同机制下调肿瘤细胞上的靶标抗原。最重要的是被靶向单价CART细胞的实体瘤被证实药效的局限性,这是因为靶标的不可接近与低表达水平。其他方法是联合scfv来靶向多种肿瘤抗原,然而,这些方法被相对大量的设计和缓慢的激活环所限制。CAR的活性和功能对于CART细胞的毒性是至关重要的,尤其是在低抗原水平的情况下。一些研究报道了CAR活性的增强提高了CART细胞的应答。通过设计,双特异性抗体表现出优于单特异性抗体的活性,因为它们可以结合同一抗原的两个不同表位。CD19和CD20双靶点CART细胞证实了有益于复发的恶性B淋巴细胞患者的一些标志,如可控的一级细胞因子释放综合征。CTA在不同癌症中得以表达,这使得它们成为免疫治疗的理想靶点。与传统靶点相比,例如EGFR, HER2, VEGF and mucin 1 (MUC1),它们也在正常组织上表达,CTA具有减小瘤外毒性的巨大潜力。尽管大多数CTA在细胞内表达,但是近期的生信研究揭示了CTA的细胞膜表达,后续的研究发现MAGE-A1在肺癌细胞系的细胞膜上表达。抗MAGE-A1 CART细胞在一个移植瘤模型上证实了肿瘤生长的抑制,该抑制是通过结合MAGE-A1阳性细胞,这验证了靶向CTA的假设。其他有CTA膜表达的,包括PRAME细胞核转录调控因子,CTA83,SP17,SLCO6A1和PLAC1,它们在膜表面与CART的结合还没有被证实。深度学习的优势和高通量筛选将会协助新一代双特异性CAR对抗典型癌症抗原的开发。6 结论和未来的展望现在治疗癌症的抗体片段可以被分成三大类:单特异性,多特异性和CAR。近期,超过30个抗体片段工程平台正在生产新型的抗体片段构型用来治疗癌症。工程方案和构型的迅速发展促使了新型抗体要的发展;因此,认真筛选合适的策略是必要的。亲和力,活性,共价键,表位互作或可接近性,稳定性和灵活性还有半衰期这些方面都需要优化来得到理想的临床药物。因此,深入理解工程策略对于设计一个合适的构型来说是十分必要的。深度学习和机器学习已经被开发应用于一系列抗体优化过程。我们应该期待通过深度学习和高通量筛选平台来探索新型抗体片段候选分子。参考文献:Lou HT, Cao XT. Antibody variable region engineering for improving cancer immunotherapy. Cancer Commun (Lond). 2022 Sep;42(9):804-827.识别微信二维码,添加生物制品圈小编,符合条件者即可加入生物制品微信群!请注明:姓名+研究方向!版权声明本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(cbplib@163.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。
免疫疗法上市批准
2023-02-13
After the FDA found multiple issues at Biocon’s manufacturing facilities in Malaysia and India, the agency has shot down Biocon and Viatris’ biosimilar to Avastin (bevacizumab).
In a
notification to the stock exchange
, biosimilar giant Biocon said the complete response letter from the FDA “informs the need for a satisfactory resolution of the observations made during the facility inspection conducted in August, 2022.” The FDA has already approved four other bevacizumab biosimilars dating back to 2017.
The statement also said that the company has submitted a Corrective and Preventive Action (CAPA) plan to the FDA, and Biocon is “confident” in “addressing the observations within the stipulated timeframe. The CRL did not identify any outstanding scientific issues with the dossier.”
Biocon popped up on the FDA’s radar in
August 2022
when the agency inspected the company’s three manufacturing facilities: two in Bengaluru, India, and one in Johor, Malaysia, as part of pre-approval inspections for several biosimilars, including bevacizumab.
Bevacizumab is used to treat several forms of cancer and is the
active compound
in Roche/Genentech’s Avastin cancer therapy.
But there were problems: in Bengaluru, procedures to prevent microbial contamination of products were not being followed, and the facility couldn’t prevent contamination of products or equipment by environmental conditions.
The FDA also noted that when there were deviations from written procedures and “laboratory mechanisms,” those deviations weren’t recorded. Also, the oversight of GMP manufacturing and laboratory operations was “inadequate.”
An inspection at a second Bengaluru facility found contamination and inadequate prevention of contamination.
In Malaysia, procedures meant to prevent microbial contamination weren’t being followed, including cleaning procedures.
In 2021, Biocon and the FDA had another run-in when one of its subsidiaries in Malaysia had an unqualified distribution system, inadequate aseptic monitoring and leaky drains.
Viatris and Biocon have partnered up on several other biosimilars including trastuzumab and pegfilgrastim.
In Feb. 2022
, Biocon announced it would buy out Viatris’ biosimilars business.
生物类似药
100 项与 贝伐珠单抗生物类似药(Vesselon, Inc.) 相关的药物交易
登录后查看更多信息
外链
| KEGG | Wiki | ATC | Drug Bank |
|---|---|---|---|
| - | - | - |
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 神经母细胞瘤 | 临床前 | - | - |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
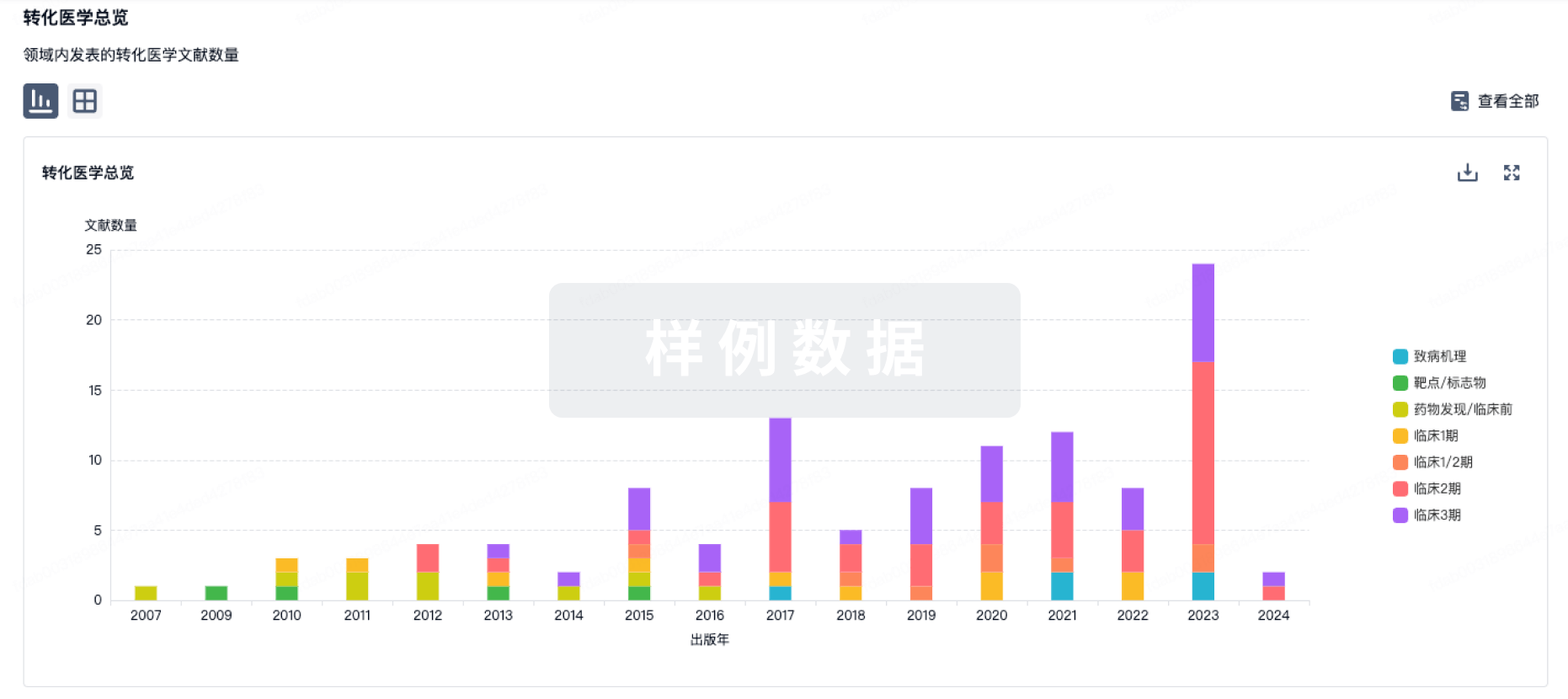
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用