预约演示
更新于:2025-03-29
AZX-100
更新于:2025-03-29
概要
基本信息
在研机构- |
最高研发阶段无进展临床2期 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |

登录后查看时间轴
结构/序列
分子式C68H114N21O18P |
InChIKeyJMPLOIHIUIQSLT-IAXAQHFISA-N |
CAS号664966-92-1 |
Sequence Code 898872

来源: *****
关联
3
项与 AZX-100 相关的临床试验NCT00892723
A Pilot Phase 2a Blinded, Placebo Controlled, Multicenter Parallel Group, Dose Ranging Study to Evaluate the Safety and Preliminary Efficacy of Additional Doses of AZX100 Drug Product Following Excision of Keloids
NCT00825916
A Pilot Phase 2a Blinded, Placebo Controlled, Multicenter Parallel Group, Dose Ranging Study to Evaluate the Safety and Preliminary Efficacy of AZX100 Drug Product Following Excision of Keloids
NCT00811577
A Phase 2a Double Blind, Placebo Within-Patient Controlled, Multi-Center Dose Ranging Study to Evaluate the Safety and Preliminary Efficacy of AZX100 Drug Product in Trocar Sites of Arthroscopic Shoulder Surgery Patients
100 项与 AZX-100 相关的临床结果
登录后查看更多信息
100 项与 AZX-100 相关的转化医学
登录后查看更多信息
100 项与 AZX-100 相关的专利(医药)
登录后查看更多信息
4
项与 AZX-100 相关的文献(医药)2010-07-01Journal of pharmaceutical sciences3区 · 医学
Internalization and Intracellular Trafficking of a PTD-Conjugated Anti-Fibrotic Peptide, AZX100, in Human Dermal Keloid Fibroblasts
3区 · 医学
Article
作者: Robert Roberson ; David Lowry ; Christopher C. Smoke ; Michael R. Sheller ; Joyce Cheung-flynn ; Charles R. Flynn ; Colleen M. Brophy
A challenge in advanced drug delivery is selectively traversing the plasma membrane, a barrier that prohibits the intracellular delivery of most peptide and nucleic acid-based therapeutics. A variety of short amino acid sequences termed protein transduction domains (PTDs) first identified in viral proteins have been utilized for over 20 years to deliver proteins nondestructively into cells, however, the mechanisms by which this occurs are varied and cell-specific. Here we describe the results of live cell imaging experiments with AZX100, a cell-permeable anti-fibrotic peptide bearing an "enhanced" PTD (PTD4). We monitored fluorescently labeled AZX100 upon cell surface binding and subsequent intracellular trafficking in the presence of cellular process inhibitors and various well-defined fluorescently labeled cargos. We conclude that AZX100 enters cells via caveolae rapidly, in a manner that is independent of glycoconjugates, actin/microtubule polymerization, dynamins, multiple GTPases, and clathrin, but is associated with lipid rafts as revealed by methyl-beta-cylodextrin. AZX100 treatment increases the expression of phospho-caveolin (Y14), a critical effector of focal adhesion dynamics, suggesting a mechanistic link between caveolin-1 phosphorylation and actin cytoskeleton dynamics. Our results reveal novel and interesting properties of PTD4 and offer new insight into the cellular mechanisms facilitating an advanced drug delivery tool.
2010-03-01Journal of neurosurgery1区 · 医学
Treatment with transducible phosphopeptide analogues of the small heat shock–related protein, HSP20, after experimental subarachnoid hemorrhage: prevention and reversal of delayed decreases in cerebral perfusion
1区 · 医学
Article
作者: Head, Geoffrey A. ; Shaver, Ellen G. ; Macomson, Samuel ; Winger, Julia ; Brophy, Colleen M. ; Harris, Valerie A. ; Furnish, Elizabeth J.
Object:
Delayed vasospasm is a significant cause of morbidity and mortality after subarachnoid hemorrhage (SAH). Proteomic therapeutics offers a new modality in which biologically active proteins or peptides are transduced into cells via covalent linkage to cell permeant peptides (CPPs). The hypothesis of this study was that either intrathecal or intravenous delivery of a phosphopeptide mimetic of the small heat shock–related protein, HSP20, linked to a CPP, would inhibit delayed decreases in cerebral perfusion after experimental SAH in a rat model.
Methods:
This study was conducted in 3 parts: 1) prevention and 2) reversal of delayed decreases in cerebral perfusion via either intrathecal or intravenous administration of a CPP linked to phosphopeptide mimetics of HSP20 (AZX100) and 3) determining the effect of intravenous administration of AZX100 on blood pressure and heart rate. Subarachnoid hemorrhage was induced in rats by endovascular perforation. Subsequently, AZX100 was administered intrathecally via a cisternal catheter or intravenously. Cerebral perfusion was determined by laser Doppler monitoring. Blood pressure was monitored by telemetry in a separate group of naïve animals treated with AZX100 for 24 hours.
Results:
The maximal decrease in cerebral perfusion occurred 3 days after SAH. Cisternal administration of AZX100 (0.14–0.57 mg/kg) 24 hours after hemorrhage prevented decreases in cerebral perfusion after SAH. Animals receiving lower doses of AZX100 (0.068 mg/kg) or a scrambled sequence of the active HSP20 peptide linked to CPP developed decreases in cerebral perfusion similar to those seen in control animals. Intravenous administration of AZX100 (1.22 mg/kg) 24 hours after hemorrhage prevented the decreases in cerebral perfusion seen in the controls. Intravenous administration (0.175 mg/kg and 1.22 mg/kg) of AZX100 on Days 2 and 3 after SAH reversed decreases in cerebral perfusion as early as Day 3. There was no impact of AZX100 on blood pressure or heart rate at doses up to 2.73 mg/kg.
Conclusions:
Cisternal administration of AZX100 24 hours after hemorrhage prevented decreases in cerebral perfusion. Intravenous administration of AZX100 also prevented and reversed decreases in cerebral perfusion at doses that did not induce hypotension. Transduction of biologically active motifs of downstream regulators like HSP20 represents a potential novel treatment for SAH.
2009-03-01The Journal of investigative dermatology1区 · 医学
Cell Permeant Peptide Analogues of the Small Heat Shock Protein, HSP20, Reduce TGF-β1-Induced CTGF Expression in Keloid Fibroblasts
1区 · 医学
Article
作者: Elizabeth J. Furnish ; Colleen M. Brophy ; Alyssa Panitch ; Luciana B. Lopes ; Patricia Ashby ; Padmini Komalavilas ; Charles R. Flynn ; Adam Hansen ; Michael T. Longaker ; George P. Yang ; Daphne P. Ly
A growing body of evidence suggests the involvement of connective tissue growth factor (CTGF) in the development and maintenance of fibrosis and excessive scarring. As the expression of this protein requires an intact actin cytoskeleton, disruption of the cytoskeleton represents an attractive strategy to decrease CTGF expression and, consequently, excessive scarring. The small heat-shock-related protein (HSP20), when phosphorylated by cyclic nucleotide signaling cascades, displaces phospho-cofilin from the 14-3-3 scaffolding protein leading to activation of cofilin as an actin-depolymerizing protein. In the present study, we evaluated the effect of AZX100, a phosphopeptide analogue of HSP20, on transforming growth factor-beta-1 (TGF-beta1)-induced CTGF and collagen expression in human keloid fibroblasts. We also examined the effect of AZX100 on scar formation in vivo in dermal wounds in a Siberian hamster model. AZX100 decreased the expression of CTGF and type I collagen induced by TGF-beta1, endothelin, and lysophosphatidic acid. Treatment with AZX100 decreased stress fiber formation and altered the morphology of human dermal keloid fibroblasts. In vivo, AZX100 significantly improved collagen organization in a Siberian hamster scarring model. Taken together, these results suggest the potential use of AZX100 as a strategy to prevent excessive scarring and fibrotic disorders.
100 项与 AZX-100 相关的药物交易
登录后查看更多信息
外链
| KEGG | Wiki | ATC | Drug Bank |
|---|---|---|---|
| - | - | - |
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 瘢痕疙瘩 | 临床2期 | 美国 | 2009-03-01 | |
| 瘢痕 | 临床2期 | 美国 | 2009-01-01 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
登录后查看更多信息
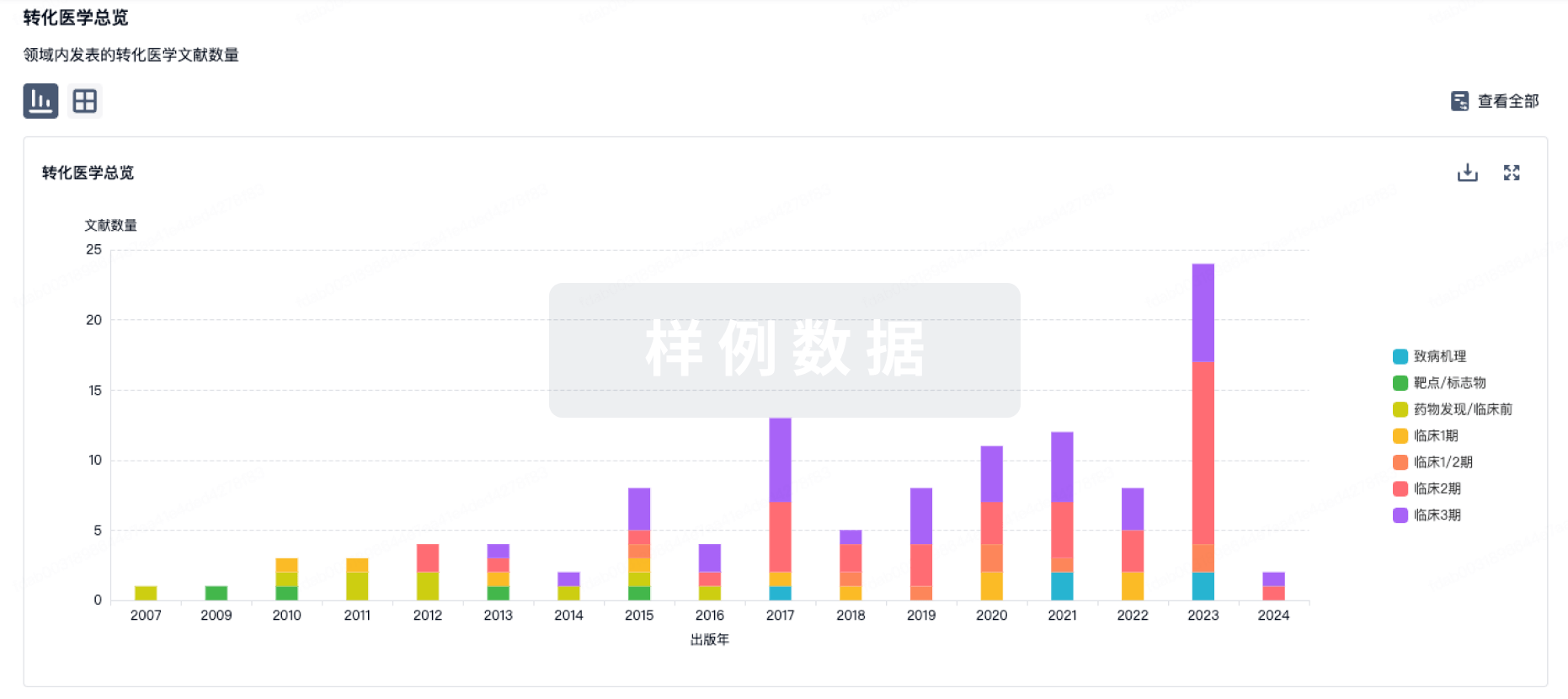
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用