预约演示
更新于:2025-04-06
SOT-102
更新于:2025-04-06
概要
基本信息
原研机构 |
在研机构- |
最高研发阶段终止临床1/2期 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |
登录后查看时间轴
结构/序列
使用我们的ADC技术数据为新药研发加速。
登录
或

关联
1
项与 SOT-102 相关的临床试验NCT05525286
A Multicentric Phase 1/2 Trial to Evaluate the Safety and Efficacy of SOT102 as Monotherapy and in Combination With Standard of Care Treatment in Patients With Gastric and Pancreatic Adenocarcinoma
This trial will assess the MTD and RP2D of SOT102 administered as monotherapy (Part A) and in combination with first-line SoC treatment (nab-paclitaxel/ gemcitabine; Part B) and efficacy of SOT102 administered as monotherapy (Part C) and in combination with first-line SoC treatment (Part D) in patients with advanced or metastatic pancreatic adenocarcinoma.
开始日期2022-03-31 |
申办/合作机构 |
100 项与 SOT-102 相关的临床结果
登录后查看更多信息
100 项与 SOT-102 相关的转化医学
登录后查看更多信息
100 项与 SOT-102 相关的专利(医药)
登录后查看更多信息
44
项与 SOT-102 相关的新闻(医药)2024-12-04
·药时代
扫码报名2024肝病新药联盟年会暨减重新药论坛!
前言
胃癌是全球最常见的恶性肿瘤之一,表现出显著的分子和表型异质性。2020年胃癌新增病例1089103例,死亡768793例,是全球第五大常见癌症和第四大癌症死亡原因。胃癌的流行病学分布因性别和地理区域而异,男性的发病率是女性的两倍,东亚和东欧的发病率更高。胃癌通常可以根据两个解剖亚型分类,此外也包括不同的组织学和分子亚型。
胃癌发生遗传畸变的积累,包括生长因子和/或受体的表达、DNA损伤反应的改变和基因组稳定性的丧失,这些表型为靶向治疗提供了各种靶点。目前癌症治疗的有效靶点包括HER2、CLDN18.2、FGFR、VEGFR、TROP2、PD-L1和Dickkopf-related protein 1(DKK1)。靶向治疗采用小分子激酶抑制剂(阿帕替尼、奈拉替尼、吡咯替尼)、单克隆抗体(曲妥珠单抗、帕妥珠单抗)、双特异性抗体(ZW25、JSKN003)、CAR-T疗法和抗体偶联药物(ADC)。
ADC是由靶向特异性抗原的单克隆抗体与小分子细胞毒性药物通过连接子链接而成,兼具传统小分子化疗的强大杀伤效应及抗体药物的肿瘤靶向性。ADC对抗原的识别导致ADC通过内吞途径进入细胞内,通过溶酶体降解后,有效载荷以生物活性形式释放并发挥作用,导致癌细胞死亡。目前,全球已有100多种候选ADC药物进行临床试验,其中15种ADC已获批上市。其中,DS-8201a、RC48和IMMU-132已被批准用于晚期胃癌治疗。因此,开发新的ADC药物和治疗模型很重要,它们可能为选择有限的晚期胃癌患者提供新的可能性。
ADC在胃癌的治疗靶点
目前临床上批准用于胃癌治疗的靶向人表皮生长因子受体2(HER2)的ADC可能无法完全满足患者的治疗需求。因此,越来越多的新靶点正在临床试验中被探索和使用。EGFR, HER3,claudin18.2、GCC、TROP2和SLC44A4均在胃癌患者中过表达,已成为ADC研究的热门靶点。
HER2
HER2作为胃癌最早、最成熟的生物标志物,一直是ADC靶向治疗的重点研究对象。T-DM1于2013年获得美国食品药品监督管理局(FDA)批准,用于治疗HER2-阳性转移性乳腺癌。然而,GATSBY研究表明,与紫杉烷相比,T-DM1对HER2阳性的晚期胃癌患者的OS和无进展生存期(PFS)没有益处,这可能是由于这些癌症中的HER2异质性。因此,鉴定胃癌中的异质性表达可能是下一步的研究方向。
与T-DM1相比,trastuzumab
deruxtecan(DS-8201a,T-DXd)由于其较高的膜通透性而表现出强大的旁观者效应,可以靶向低HER2表达和HER2异质性胃癌患者。临床前实验表明,DS-8201a通过抑制ADCC和Akt磷酸化在HER2阳性NCI-N87模型中表现出抗肿瘤作用。此外,DS-8201a与吡咯替尼联合使用可促进泛素化,并增强T-DXd在胃癌细胞中的吸收。RC48(disitamab vedotin),另一种ADC,于2021年获得中国国家医疗产品管理局的批准,在胃癌患者中显示出良好的抗肿瘤活性。目前正在进行进一步测试,以评估其治疗胃癌的疗效(NCT05313906、NCT05241899、NCT05980481)。
此外,一些未上市的ADC也显示出巨大的潜力,如ARX788、PF-06804103、XMT-1522和MEDI4276等,这些ADC也正在进行临床试验,以评估胃癌患者的剂量和组合模式。
EGFR和HER3
MRG003是中国首个测试的EGFR靶向ADC,目前正在参与一项II期临床研究(NCT05188209),评估其治疗晚期胃癌的疗效和安全性。EV20/NMS-P945在涉及猴子的药代动力学和毒理学研究中显示出良好的半衰期和稳定性。此外,它在体外的各种肿瘤细胞系中表现出靶向依赖性细胞毒性活性,包括胰腺癌、前列腺癌和胃癌。AMT-562在源自胃癌患者的低HER3表达细胞系和患者来源的肿瘤异种移植物(PDX)模型中产生强烈而持久的抗肿瘤反应。BL-B01D1,一种靶向EGFR和HER3双特异性抗体偶联形成的新型ADC,正在与SI-B003一起试验,用于治疗胃肠道肿瘤,包括胃癌(NCT06008054)。
CLDN18.2
CLDN18.2是一种参与细胞旁紧密连接结构的重要膜蛋白,在正常生理条件下仅在胃黏膜的分化上皮细胞中表达。该蛋白在近60%的胃癌患者中持续稳定表达。2021年3月,CMG901(AZD0901)被FDA批准用于针对晚期实体瘤(胃癌和胰腺癌)的I期临床研究(NCT04805307),在89名患者中实现了33%的客观缓解率(ORR)和70%的疾病控制率(DCR)。另一个靶向Claudin18.2的ADC EO-3021(SYSA1801)在17名胃癌患者(NCT05009966)中显示出疗效,ORR为47.1%,DCR为64.7%。
此外,其他ADC正处于早期研究阶段,如TORL-2-307和LM-302,它们正在研究CLDN18.2阳性晚期胃肠道癌症患者的临床疗效,无论是作为单一药物(NCT05156866)还是与toripalimab联合使用(NCT05934331)。SOT102(NCT05525286)和CPO102(NCT05043987)已在胃癌和胃食管交界处癌患者中显示出初步疗效。
GCC
GCC属于受体鸟苷酸环化酶家族的成员,在胃肠道的液体和离子稳态中起着关键作用,在原发性和转移性胃肠道癌症中高表达。其表达水平在结直肠癌中达到约95%,在胰腺癌、胃癌和食管癌中达到60-70%。
在评估TAK-264(NCT01577758)的一项人类I期研究中,在胃癌、胃食管癌和胰腺癌中观察到了初步临床疗效。然而,随后的II期研究(NCT02202759)显示,使用TAK-264治疗的一小部分患者的临床益处有限。尽管靶向GCC的ADC尚未达到最佳治疗效果,但由于GCC在胃癌中的高表达,它们继续显示出治疗晚期胃癌的巨大潜力。
TROP2
TROP2在多种肿瘤中高表达,并可促进肿瘤细胞增殖、侵袭、转移和扩散。TROP2的高表达通常与生存期缩短和预后不良有关。
IMMU-132(sacituzumab
govitecan)和SKB264都由可切割的连接子和拓扑异构酶I抑制剂组成。IMMU-132在胃癌的活性已在I/II期临床试验(NCT01631552)中报道。一项正在进行的研究(NCT04152499)评估了SKB264作为单一疗法,可能使局部晚期不可切除或转移性实体瘤对标准疗法产生耐药性的患者受益,包括胃腺癌和胃食管交界处腺癌。此外,DS-1062在几种表达TROP2的异种移植物模型中显示出抗肿瘤作用,包括NCI-N87模型。其在晚期/转移性胃癌患者中的试验(NCT05489211)正在进行中。
SLC44A4
SLC44A4通常在分泌上皮细胞的顶面上表达,现已被确定为大多数胰腺癌和胃癌中的新型细胞表面靶标。靶向SLC44A4的ADC药物ASG-5ME的临床研究(NCT01166490)显示,15名胃癌患者接受1.2 mg/kg的最大耐受剂量, DCR为47%。
ADC胃癌的联合治疗
联合治疗现在是肿瘤学临床前和临床研究的核心焦点。将ADC与各种疗法相结合,有利于改善实体瘤患者的预后。
ADC联合单克隆抗体治疗
几项正在进行的临床试验表明,ADC与mAbs联合治疗可提高各种类型胃癌的疗效。一项纳入30名晚期胃癌患者的试验(CTR20639)表明,与曲妥珠单抗单药治疗相比,ARX788联合治疗可显著延长患者的PFS和OS分别达4.1个月和10.7个月。在另一项临床试验(NCT04014075)中,证明DS-8201a可以治疗接受曲妥珠单抗治疗的晚期胃癌患者,从而导致更高的ORR和更长的OS。在联合治疗取得有希望的结果后,DESTINY-Gastric04研究(NCT04704934)目前正在评估在接受曲妥珠单抗治疗后,将T-DXd或ramucirumab和紫杉醇(Ram+PTX)联合治疗HER2阳性的胃癌或胃食管交界腺癌患者的疗效和安全性。
ADC联合免疫疗法
目前,阻断免疫检查点抑制剂已成为增强癌症患者抗肿瘤反应的一种有前景的方法。研究表明,ADC有效载荷直接诱导树突细胞活化和成熟,引发免疫原性细胞死亡。此外,ADC结构内的单克隆抗体可以上调PD-1/PD-L1的表达,增强免疫细胞浸润,促进树突状细胞的抗原呈递,最终提高PD-1/PD-L1抑制剂的疗效。
在一项涉及HER2表达的局部晚期或转移性实体瘤患者的试验中,RC48与JS001(PD-1抗体)联合使用(NCT04280341)。本试验报告胃癌组患者的ORR为43%,DCR为75%,反应持续时间为5.1个月,PFS为6.2个月,mOS为16.8个月。此外,T-DXd已被证明可以增加肿瘤浸润CD8+T细胞的数量,并增强肿瘤细胞中PD-L1和MHC-I的表达。该药物目前正在胃癌患者中单独或与durvalumab(一种PD-L1抗体)联合使用,以评估其安全性、耐受性、药代动力学、免疫原性和初步抗肿瘤疗效(NCT04379596)。
ADC联合其它药物
对于HER2阳性的晚期胃癌,抗HER2靶向治疗和化疗的联合治疗是标准的一线治疗,而基于ADC的联合治疗目前正在积极研究中。在RC48-C008研究(NCT03556345)中,RC48被用于先前接受化疗的局部晚期或转移性胃癌患者,从而形成了联合治疗方法。研究结果表明,ORR为24.8%,PFS为4.1个月,mOS为7.9个月。
药物的选择会影响ADC与靶向表面抗原的结合。例如,pyrotinib显著诱导HER2受体的内化,增强癌症细胞对T-Dxd的摄取。洛伐他汀是一种降胆固醇药物,可暂时增加细胞表面HER2水平,增强HER2-ADC结合和内化。WEE1是一种调节肿瘤细胞分裂的蛋白质,强效WEE1抑制剂adavosterib和T-DXd的组合显著增加了抗肿瘤活性,这是因为Wee1激酶抑制剂在G2期阻滞期间绕过了细胞修复复制应激诱导的损伤的能力,导致DNA损伤积累和随后的有丝分裂破坏。
小结
ADC在胃癌治疗中备受期待,目前上市的DS-8201a、RC48和IMMU-132已成为晚期胃癌患者的二线和后线治疗选择。然而,很少有胃癌患者存活下来接受二线或二线治疗,因此,有必要进一步探索新的ADC药物和联合疗法作为一线和二线治疗。
此外,确定最佳的联合治疗和克服耐药性的方法应在未来的研究中加以解决。偶联技术和新型连接子或有效载荷的增强可以为下一代ADC的胃癌治疗提供思路。鉴于肿瘤环境的复杂性,双特异性ADC可能潜在地促进双重靶点的协同内吞作用,并提高有效载荷进入癌细胞的效率,这也有利于克服耐药性。未来ADC在胃癌和其它实体瘤治疗中应有更广阔的发展前景。
参考文献:
1.Antibody-drug
conjugates in gastric cancer: from molecular landscape to clinical strategies. Gastric
Cancer.2024 Jul 4.
封面图来源:123rf
抢先剧透!2024肝病新药联盟年会暨减重新药论坛(第三轮通知)
2024-12-03
肝病重磅药,正式撤市!
2024-12-02
【新药进展】全球乙肝新药进展(更新至2024年11月,独家整理)
2024-12-01
版权声明/免责声明
本文为授权转载文章。
本文仅作信息交流之目的,不提供任何商用、医用、投资用建议。
文中图片、视频、字体、音乐等素材或为药时代购买的授权正版作品,或来自微信公共图片库,或取自公司官网/网络,部分素材根据CC0协议使用,版权归拥有者,药时代尽力注明来源。
如有任何问题,请与我们联系。
衷心感谢!
药时代官方网站:www.drugtimes.cn
联系方式:
电话:13651980212
微信:27674131
邮箱:contact@drugtimes.cn
点击这里,报名参加行业盛会!
抗体药物偶联物细胞疗法申请上市免疫疗法临床1期
2024-11-07
PRAGUE & BASEL, Switzerland & BOSTON--(
BUSINESS WIRE
)--
SOTIO Biotech
, a clinical-stage immuno-oncology company owned by PPF Group, today announced data supporting SOT201, its next-generation PD-1-targeting immunocytokine. The company also reported advancements in its BOXR cell therapy platform, introducing an innovative chimeric PGC-1α transgene to boost CAR T cell efficacy in patients with solid tumors. SOTIO will be presenting three posters highlighting these advancements at the 2024 Society for Immunotherapy of Cancer Meeting, taking place November 6–10 in Houston, TX, U.S.
SOT201 is a PD-1-targeted and cis-acting attenuated IL-15 agonist designed to preferentially activate PD-1
+
CD8
+
T cells, inducing superior anti-tumor effects and reinvigorating exhausted CD8
+
T cells in PD-1 sensitive and resistant tumors. The VICTORIA-01 study is a Phase 1, open-label, dose escalation trial that aims to assess the safety, tolerability, and preliminary efficacy of SOT201 as a monotherapy for adults with advanced unresectable or metastatic solid tumors (
NCT06163391
). This trial is currently enrolling patients across six sites in the U.S., Belgium, Spain, and the Czech Republic. Four patients have been treated so far and the treatment was well tolerated.
SOTIO Chief Scientific Officer Martin Steegmaier, Ph.D., noted, “
SOT201 demonstrates a superior ability to reinvigorate exhausted tumoral CD8
+
T cells with a high cytotoxicity and minimal cellular exhaustion compared to the related cytokine PD1-IL2v. These data reinforce SOT201’s reduced off-target interactions and more durable anti-tumor efficacy
in vivo
, underscoring its potential to address current limitations in anti-PD-1 therapies, as we continue to enroll patients in the VICTORIA-01 study.”
The third poster highlights a preclinical study of a chimeric PGC-1α transgene that enhances CAR T cell activity. Chimeric PGC-1α transduced cells displayed fewer dysfunctional mitochondria and improved glucose uptake compared to CAR T cell controls. “
Furthermore, the chimeric PGC-1α enhanced CAR T anti-tumor efficacy with no overt signs of toxicity, suggesting that co-expression of CAR and the chimeric PGC-1α is a promising approach to improving CAR T cell efficacy in solid tumors,” added Dr. Steegmaier.
Presentation materials will be available
here
on Sunday, November 10, after the conference concludes.
About SOTIO Biotech
SOTIO Biotech (SOTIO) is shaping the future of cancer immunotherapies by translating compelling science into patient benefit. The SOTIO pipeline includes SOT102, a next-generation Claudin-18.2-targeted antibody-drug conjugate which entered the clinic in 2022; BOXR1030, a metabolically-enhanced CAR-T cell therapy targeting GPC3-expressing tumors as well as other molecules approaching clinical stage such as SOT201, our next-generation PD-1-inhibiting immunocytokine. SOTIO is a member of the PPF Group. For more information, please visit the company’s website at
www.sotio.com
.
SOTIO is a registered trademark of SOTIO Biotech a.s. in selected countries.
免疫疗法细胞疗法临床1期AACR会议
2024-11-02
·摩熵医药
注:本文不构成任何投资意见和建议,以官方/公司公告为准;本文仅作医疗健康相关药物介绍,非治疗方案推荐(若涉及),不代表平台立场。任何文章转载需要得到授权。
胃癌作为一种常见的恶性肿瘤,其治疗手段和药物研发一直是医药市场的关注焦点。随着医疗技术的不断进步和人们对健康需求的日益增长,胃癌市场的治疗方法和药物研发呈现出蓬勃发展的态势。其中,CLDN18.2靶点的发现为胃癌治疗带来了新的机遇。
本文基于摩熵咨询(原药融咨询)最新权威发布的《胃癌药物市场研究专题报告》部分精彩内容,深入剖析胃癌治疗市场的最新趋势,特别聚焦于CLDN18.2靶点相关药物的研发动态,旨在为读者呈现该领域的前沿进展与广阔前景。
01
CLDN18.2有望成为
胃癌靶向治疗理想靶点
紧密连接蛋白(Claudin,CLDN)在邻近细胞间可形成紧密通道,调控离子、溶质等物质流通。CLDN18.2在胃癌、食管癌等恶性肿瘤中特异性高表达,且肿瘤细胞因增殖快、易侵袭转移等特性丧失紧密连接结构,导致其表面的CLDN18.2分子表位更易暴露出来。
因此CLDN18.2在众多潜力型靶点中脱颖而出,有望成为胃癌靶向治疗理想靶点。目前,CLDN18.2可开发成多种不同类型药物,包括单抗、双抗、ADC、CAR-T,为胃癌治疗提供更多选择。
ICH是目前应用最广泛的检测手段,不同研究中CLDN18.2的表达值存在较大差异,不同种族之间阳性率差异性较高。在一项中国人群的研究中,胃印戒细胞癌CLDN18.2的阳性率高达95.2%。
02
CLDN18.2靶向药物
市场现状及展望
(1)靶向CLDN18.2的单克隆抗体药物
Zolbetuximab(佐妥昔单抗)是全球第一个靶向Claudin18.2(CLDN18.2)的IgG1单克隆抗体,其通过与肿瘤细胞表面CLDN18.2结合从而激活抗体依赖的细胞介导的细胞毒性作用(ADCC)和补体依赖的细胞毒性(CDC)效应诱导癌细胞死亡。
2024年3月,安斯泰来宣布其创新药物佐妥昔单抗(Zolbetuximab)获日本厚生劳动省批准,用于治疗不可切除晚期或复发性CLDN18.2阳性胃癌。目前,我国国家药监局药审中心已受理其上市申请。
据摩熵医药(原药融云)数据库显示,目前全球已进入临床阶段的重点CLDN18.2单抗产品有近20款,其中进入III期临床及以上的共有4款,包括安斯泰来的Zolbetuximab、明济生物的FG-M108、创胜集团的osemitamab、奥赛康药业的ASKB-589。;处于II期临床阶段的有再鼎医药的ZL-1211、礼新医药的LM-102。
Osemitamab (TST001) 是创盛集团自主研发的一种高亲和力的靶向Claudin18.2的人源化单克隆抗体,具有增强的抗体依赖性细胞毒性(ADCC)和补体依赖性细胞毒性(CDC)活性,在异种移植试验中显示出强大的抗肿瘤活性。
中国和美国均一直在进行Osemitamab的临床试验(TranStar101/NCT04396821, TranStar102/NCT04495296)。美国FDA已授予Osemitamab用于治疗胃癌或胃食管结合部腺癌和胰腺癌患者的孤儿药资格认定。
2024年5月,创盛集团公布了Osemitamab联合纳武利尤单抗和CAPOX作为晚期胃或胃食管结合部腺癌一线治疗的I/IIa期G队列研究数据。该项研究的患者未经过CLDN18.2和PD-L1 CPS表达筛选入组,研究结果显示三联疗法取得令人鼓舞的疗效,特别在CLDN18.2高/中表达的患者群体中(无论PD-L1 CPS表达水平如何)尤为显著。
ASKB589为奥赛康自主研发的靶向CLDN18.2的重组人源化单克隆抗体药物。作为第二代抗CLDN18.2人源化单克隆抗体,ASKB589在对CLDN18.2的亲和力和特异性方面显著优于第一代产品。
ASKB589全球研发现状截图来源:摩熵医药全球药物研发数据库
摩熵医药数据库显示,2023年10月,ASKB589注射液获CDE批准在中国开展III期关键性临床试验。其在II期临床试验中的优异表现,包括高的客观缓解率和疾病控制率,以及可接受的安全性和耐受性,均表明这种新疗法在治疗CLDN18.2阳性胃癌及胃食管交界处腺癌方面具有重大的临床应用价值。
(2)靶向Claudin18.2的双抗药物
双抗药物可以同时特异性结合2个抗原或抗原表位的人工抗体,已成为近几年新药研发的热点。
目前关于CLND18.2双抗的另一个靶点大多针对免疫细胞相关分子设计,旨在发挥抗肿瘤免疫作用。主要为CD3/CLDN18.2、CD47/CLDN18.2、4-1BB/Claudin18.2、PD-1/CLDN18.2这几类。
研究表明CD47-SIRPα信号轴是是癌症的先天免疫检查点,抗CD47和抗SIRPα可以阻断CD47-SIRPα抑制信号并促进巨噬细胞对肿瘤细胞的吞噬。
摩熵医药数据库显示,目前已有多个CD47/CLDN18.2双抗进入临床阶段。如凡恩世生物的PT-886已进入Ⅱ期临床,宝船生物/三优生物的BC-007、康方生物的AK-132、尚健生物的SG-1906也已进入Ⅰ期临床。
(3)靶向CLDN18.2 的ADC药物
ADC药物是最近几年抗肿瘤领域非常火热的领域,由具有生物活性的细胞毒性药物和mAb通过化学键连接而成,并由mAb搭载细胞毒性药物进入特定细胞发挥肿瘤杀伤作用。
胃癌领域目前研究最多的ADC药物主要聚焦在HER2靶点上,但靶向CLDN18.2的ADC药物也在陆续进入临床阶段。
部分ADC药物由CLDN18.2单抗与单甲基澳瑞他汀E结合而成,如阿斯利康的CMG-901、百时美施贵宝/礼新医药的LM-302、石药集团的COP-102;部分ADC药物由CLDN18.2单抗与DNA拓扑异构酶抑制剂结合而成,如恒瑞医药的SHR-A1904、NBE的SOT-102等。
(4)CLDN18.2靶向的CAR-T疗法
CAR-T是通过基因工程手段,在T细胞的细胞膜上嵌合某种特定肿瘤抗原受体基因,形成修饰的T细胞,进而可特异性识别和结合肿瘤细胞表面的抗原,并引导T细胞杀伤特定的肿瘤细胞。
Satricabtagene autoleucel是目前研发进展最快的CLND18.2靶向的CAR-T疗法,也是在胃癌领域取得较好结果的CAR-T。在中国人群中开展了Ⅰ期临床试验,中期分析结果显示,CT041治疗既往接受过治疗的CLDN18.2阳性消化系统肿瘤患者的耐受性良好,总体人群ORR为48.6%,DCR为73.0%;在胃癌患者中,ORR为57.1%,DCR为75.0%,6个月OS率为81.2%。
03
胃癌市场的未满足临床
需求与未来展望
尽管胃癌治疗手段不断丰富,但晚期胃癌的整体预后仍不佳,单纯化疗效果提升进入瓶颈期,靶向药物选择有限,免疫单药在整体人群中疗效不佳,PD-1单抗联合化疗已成为一线治疗优先选择,但免疫联合策略有待进一步优化和提升。
基于已公布的临床数据,CLDN18.2有望成为胃癌的第二个精准治疗靶点,左妥昔单抗已验证其胃癌成药性,国产药物研发势头正盛。2024 CSCO胃癌指南也推荐患者在术后进行IHC检测,以评估CLDN18.2表达情况。在CLDN 18.2靶向药物获批上市后,晚期复发患者能够根据既往检测结果及时接受相应靶向治疗。一线治疗方案预计为CLDN18.2单抗联合化疗联合/不联合PD-1单抗。
目前,国内有多款药物正处于胃癌适应症的申请上市阶段,其中包括康方生物的卡度尼利单抗、阿斯利康的德曲妥珠单抗、和记黄埔医药的呋喹替尼以及安斯泰来的左妥昔单抗。这些新药为胃癌治疗带来了新的希望。
结语
随着医学研究的深入,胃癌治疗领域正迎来一场革命。CLDN18.2靶点的发现和相关药物的研发,不仅为胃癌患者提供了新的治疗选择,也为整个医药市场注入了新的活力。从单克隆抗体到双抗药物,再到ADC药物和CAR-T疗法,多样化的治疗手段正在逐步满足胃癌患者的临床需求。尽管挑战依然存在,但未来的治疗前景无疑是光明的。我们期待这些创新药物能够早日获批上市,为胃癌患者带来更有效的治疗方案,改善他们的生存质量和预后。
市场研究专题报告:胃癌药物完整报告领取
识别下方二维码领取
END
本文为原创文章,转载请留言获取授权
联系我们,体验摩熵医药更多专业服务
会议
合作
园区
服务
数据库
咨询
定制
服务
媒体
合作
点击阅读原文,了解更多品牌升级详情!
抗体药物偶联物细胞疗法免疫疗法临床2期临床3期
100 项与 SOT-102 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 晚期胰腺腺癌 | 临床2期 | 美国 | 2022-03-31 | |
| 晚期胰腺腺癌 | 临床2期 | 比利时 | 2022-03-31 | |
| 晚期胰腺腺癌 | 临床2期 | 捷克 | 2022-03-31 | |
| 晚期胰腺腺癌 | 临床2期 | 法国 | 2022-03-31 | |
| 晚期胰腺腺癌 | 临床2期 | 西班牙 | 2022-03-31 | |
| 胰腺腺癌 | 临床2期 | 美国 | 2022-03-31 | |
| 胰腺腺癌 | 临床2期 | 比利时 | 2022-03-31 | |
| 胰腺腺癌 | 临床2期 | 捷克 | 2022-03-31 | |
| 胰腺腺癌 | 临床2期 | 法国 | 2022-03-31 | |
| 胰腺腺癌 | 临床2期 | 西班牙 | 2022-03-31 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
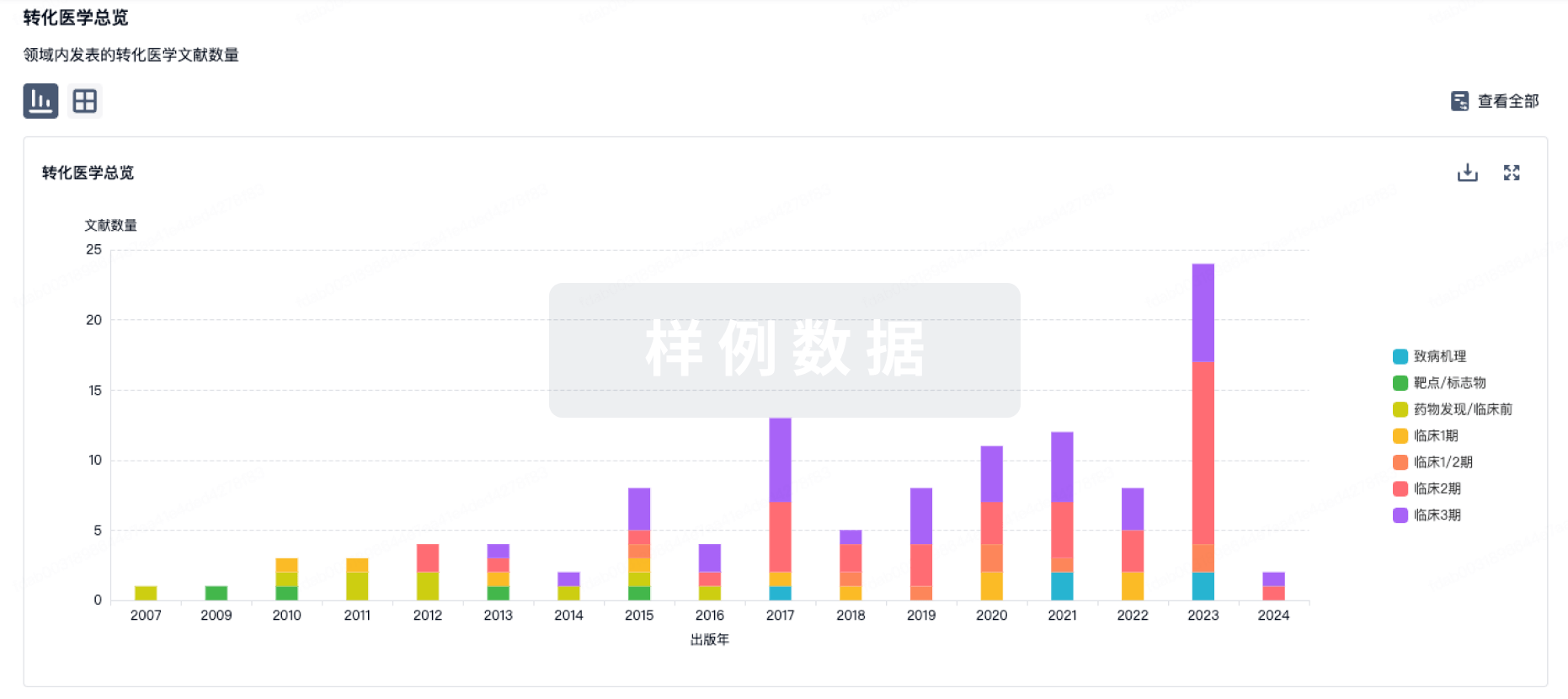
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用