预约演示
更新于:2025-04-08
CMT1A(Xili Technology )
CMT1A(溪砾科技)
更新于:2025-04-08
概要
基本信息
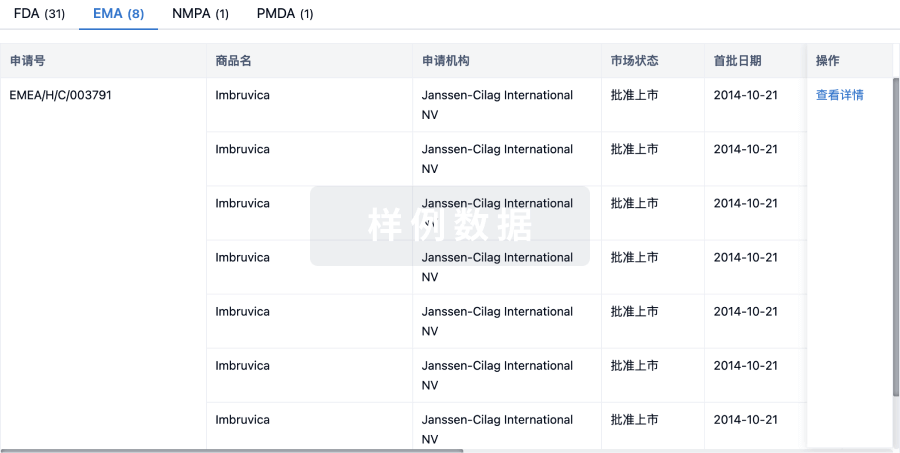
原研机构 |
在研机构 |
非在研机构- |
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)临床前 |
特殊审评- |
关联
100 项与 CMT1A(溪砾科技) 相关的临床结果
登录后查看更多信息
100 项与 CMT1A(溪砾科技) 相关的转化医学
登录后查看更多信息
100 项与 CMT1A(溪砾科技) 相关的专利(医药)
登录后查看更多信息
7
项与 CMT1A(溪砾科技) 相关的新闻(医药)2024-07-09
在生命科学的爆发年代,当CGT、蛋白组学研究突飞猛进,是时候加速与时间竞逐,用力为罕见病治疗凿开一道口子,在窄途之上打开道道“生门”。
“偶尔治愈,常常缓解,总是安慰。”
美国医生爱德华·特鲁多的墓志铭,从另一角度上体现了当前罕见病患者的治疗现状:难以治愈,甚至无法治愈,只能尽力缓解和安慰。
在中国,罕见病已成为创新、医保之外,药界最为关注的三大话题之一。探索罕见病用药,不仅是为了满足数位患者及家庭的未满足需求,更是对中国生物制药关于“创新”这一核心命题的层层拆解。因为从本质上来看,罕见病用药在研发、诊疗、支付到进院等多个环节上遇到的问题,恰是创新药面临的所有问题的集合。
2018年,中国《第一批罕见病目录》公布,从渐冻症(ALS)、腓骨肌萎缩症(CMT)、全身型重症肌无力,再到亨廷顿舞蹈病,与神经病学相关的病种多达40个。6年时间过去,这些疾病领域已取得诸多进步,但仍缺量和质的飞跃性突破,还亟需政策、法律、资本、企业等多方共同努力。其中,作为创新主体的企业,需在技术突破、药物研发上,带来新的治愈希望。
近期,E药经理人便发现了这样一笔合作:AI+小分子靶向RNA疗法研发企业ReviR溪砾科技(以下简称ReviR),与CMT研究基金会(CMT Research Foundation,CMTRF)宣布建立战略合作关系,CMTRF对ReviR进行投资,推进调节CMT1A致病基因表达的小分子靶向RNA疗法研究。公开资料显示,除了天士力医药引进的新药PXT3003,ReviR是目前首个在国内公开发布关于CMT研究的企业。
CMT,腓骨肌萎缩症,一种遗传性神经疾病,患病率约1/2500。然而,这种罕见病目前呈现“三无”特点:无任何治疗方法或治愈手段,“更多是‘对症治疗’,而非针对病因的治疗。”北医三院神经科主任樊东升这样介绍;国内针对这一适应证的临床项目相对不多,且无重大突破进展;此外,CMT并无太大的社会认知和公众关注度。
而ReviR与CMTRF的合作,旨在利用新兴RNA疗法,以AI技术助力,从基因层面切入寻求潜在的可口服疗法。这一举措,或将助力罕见病药物研发史上出现一位关键的“破冰者”。
01
CMT耐力赛上,有了新鲜“血液”
CMT,大约每10000人中就有4位患者,但其在公众中的认知度相对偏低,常存于人群里,却又好似被隐匿。
很多人易将渐冻症与CMT相混淆,主要是因为两种疾病都涉及肌肉功能的障碍和萎缩,但实际上,这两种疾病存在本质区别。
渐冻症为一种罕见但致命的神经退行性疾病,主要影响运动神经元,而CMT虽然也被列入了罕见病目录,但就其发病率而言,是一种常见的遗传性运动感觉神经病,主要侵犯周围神经,目前病因不明,分型很多,并发症也各不相同。已知超100个不同的致病基因中,CMT1型(脱髓鞘型)占据50%以上。
然而,发病率相对较高的CMT,反而更处在一个知名度和公众关注度较低、同时没有有效逆转病程的治疗方法的“尴尬”位置。
“缺乏根治手段,大多数病人诊断出来以后都无药可治,治疗上相对比较滞后。”樊东升研究神经系统疾病多年,腓骨肌萎缩症的治疗现状让他有些着急。如他所言,目前,关于CMT的治疗,主要是通过药物治疗(如B12等神经营养药物)及支持治疗,以延缓疾病的进展,如,外科矫正手术、康复治疗、心理支持等。
他坦言,这类疾病,一是亟待从源头上解决问题;二是缺乏新药研发投入,尤其在国内,有关CMT的研究整体起步较晚;三仍缺人才;四缺乏足够的“声量”;五则缺乏整个社会大环境的支持。
罗玉萍是一名腓骨肌萎缩患者。去年1月,由罗玉萍等患者及患者家属共同发起成立的千里行CMT互助之家,为患者搭建起了一个沟通交流平台,以促进医疗机构和科研单位对CMT的医学研究。站在罗玉萍及千里行CMT互助之家这一组织的第一视角,CMT患者正面临很多现实问题,典型如:
诊疗时,找不到合适的医生就诊。
公众认知上,对腓骨肌萎缩症的认识不足,就诊分类存在一定错误。
自我感知上,由于缺乏专业知识,往往难以准确判断自己的病情。基于CMT病情进展通常缓慢,患者可能在一段时间内并不能察觉到明显变化,即使有所察觉也未能及时就医。
随着时间推移,CMT患者会因腿部和手臂肌肉逐渐萎缩,导致骨骼畸形、手脚功能异常、平衡能力受限,易出现行走、跑步等困难,还可能会有感觉减退,呈现手套-袜套样感觉障碍,CMT1型可能还会出现神经性耳聋,表现为听力减退,少数患者需要依赖轮椅生活。
受访者供图
故当看到与CMTRF的这笔合作时,一个作为国内CMT药研的牵头研究者(PI),一个作为为患者发声的代表,樊东升和罗玉萍均表达了其激动与期待。
樊东升看到,在最有希望获得突破的CMT基因疗法上,国内终于有企业迈出了新的一步,这或将成为国内针对CMT研究的重要里程碑;而在罗玉萍及所有CMT患者眼中,任何可能改善症状、延缓疾病进展或提供治疗选择的新疗法,其探索都极其宝贵。
那么,ReviR究竟采取了什么样的原理及方式,在基因治疗层面寻求突破的?
02
以“可口服的基因疗法”为目标,探索“无人区”
2021年成立的ReviR,是一家结合Al技术进行靶向RNA小分子药物研发的生物科技新锐,主要针对中枢神经系统疾病、癌症和其他罕见遗传性疾病进行创新药的研发。
针对CMT这一罕见病,目前ReviR正积极探索一种与传统高侵入性基因疗法存在本质区别的治疗路径。
据悉,ReviR会开发一种小分子剪接调节剂,以降低CMT1A致病基因的过表达,从而缓解疾病症状。这种剪接剂可以通过穿越血脑屏障,有效降低致病基因蛋白表达,从基因层面阻断CMT1A的发生。与其他需要注射的疗法相比,这种药物具备可口服的优势,更加方便递送,能革新CMT1A治疗模式的同时,还有望用于CMT其他分型以及其他神经退行性疾病。
这一治疗路径,获得了CMT领域关键力量的青睐与支持。
CMTRF,由患者发起组织、募资并资助企业进行CMT新药研发。自该组织2018年成立至今,全球CMT药物研发管线增加了一倍多,已取得了诸多阶段性成果。截至目前,CMTRF已资助24个项目,其中8个已经顺利完成。在已完成的项目中,有5个项目开发出了临床候选药物,有的项目已被大型跨国药企收购。在此助力下,愈高的社会关注和企业投入也为患者们带来了更多信心。而CMTRF的成果,同时为罕见病探索提供了一个有效的借鉴。
但诚如CMTRF的创始人之一Susan Ruediger向E药经理人感慨,2018年组织成立时,全球尚未有获批的CMT治疗药物,6年时间过去,情况仍旧如此。
6年时间里,CMTRF都在助力寻找CMT领域的“更优解”。此次选择与ReviR合作,CMTRF首席执行官(CEO)Cleary Simpson便向E药经理人道出了核心原因:ReviR有望提供一种非侵入性、有效、又易于管理和给药的疗法,或将为CMT患者带来新的生活改善,而这种疗法一经突破,还可能颠覆其他分型的CMT和其他神经退行性疾病的治疗现状。
而于ReviR本身而言,这一合作能极大加快其研究进展。ReviR公司首席科学官(CSO) Paul August向E药经理人表示:“与CMTRF的合作,能为我们提供独特的资源、患者数据,以及科学和临床专家网络,极大加快我们的研究进展。”基于此,他透露预计将在2027年或2028年左右,开始进入I期临床试验,目标是对标诺华等大型药企,与之达到有效竞争。
尤其值得关注的是,针对CMT要打造的可口服基因疗法,是基于ReviR独有的AI药物研发技术平台——VoyageR而来。
VoyageR包括RNA靶点发现和小分子药物设计两大功能模块,结合自有的海量湿实验及真实世界数据进行机器学习模型自动化迭代,在广阔的RNA靶点与化合物海洋里快速筛选、高速优化,从而推进靶向RNA的小分子药物研发。
成立至今,搭建了VoyageR平台的ReviR,不仅囊获诸多明星投资机构支持,还成功与本土知名Biopharma达成了重磅战略合作。
基于VoyageR,ReviR致力于开发一系列调控剪接的小分子药物——剪接剂(SpliceR),核心在于通过影响mRNA转录本来调节基因表达,最终达到治疗效果。SpliceR属于一种新的基因治疗方式,其作用机理类似于治疗脊髓性肌萎缩症的疗法Evrysdi。但两大药物调整方式有些微不同,通俗来说,ReviR的SpliceR药物是在遗传信息中增加了一段,告诉机体停止表达蛋白质,Evrysdi则是通过调整基因来产生身体所需的重要蛋白质。
03
以AI+RNA为匙,助力探索小分子“大前沿”
ReviR在积极探索一件很新的事情。
核酸药物的探索,是一场漫长的冒险,真正的出路在于率先开发出有价值的新应用场景,并且做扎实。但这一路径,行者均称艰难。
即便mRNA技术因新冠大流行而突飞猛进,但迄今,这条技术路径上仍很难看到有明显差异化的、真正原创的产品。即使有,要么背后对疾病机理已有深入的研究,要么是基于前期多年的积累。
小核酸药物的研发,同样历经了很漫长的“痛苦期”,即便是先驱企业,被全世界发现之前,他们集体“枯坐”了超10年的冷板凳。
相较于mRNA或小核酸药物,RNA小分子药物的研发,更处在一个较早期阶段,已经进入市场的药物还很少,成功的案例,通常都出自偶然。尤其是在国内,这一类型的药物亟需开拓与纵深。但囿于更易变幻和复杂的结构,RNA小分子药物研发面临着重重困难。要对RNA小分子药物研发注入更多的“必然”,需要更好的测序技术、筛选方法和对RNA生物学、疾病机理更深入的理解等多重条件的满足。
2021年,ReviR 成立,将AI技术思路与RNA结合,基于RNA靶向小分子库,结合高通量筛选+AI+自动化实验验证干湿闭环,既能快速理解哪些成分在起作用,还能提速筛选得到临床前候选化合物,不仅在速度、效率、成本上给传统制药带来了颠覆,在疗法研究上也具备开创性意义与独有优势。
作为领先探索的AI+小分子靶向RNA疗法企业,ReviR已实现“硬件”和“软件”上的武装:一边,基于全球生物技术研发前沿趋势的判断,一个颇具差异化特点的VoyageR AI平台技术已就位;另一边,一个在药物研发、计算化学、计算生物学等领域有深厚背景和经验的团队,也已速速搭建。核心团队里,联合创始人兼CEO岳鹏曾在罗氏基因泰克、吉利德、艾伯维等MNC就职;联合创始人李阳在AI算法、生物信息学等方向上研究深入;Paul August是全球首款治疗成人丙酮酸激酶缺乏症Pyrukynd的发明人……
很多时候,AI制药企业要面临的挑战,其实来自于对生物学机理的认知。所谓创新药物的创新靶点,并非是“别人不敢做的我敢做”,或者在没有临床试验数据可以佐证的情况下敢于“杀”进去,而是对于要做的这一目标靶点,对于适应证、适用人群、联用方案等的选择,有着比同行更为深刻的理解。对应地,ReviR 的“硬核”跨学科团队,是其作为一家AI制药企业难得的竞争力。
基于此,ReviR在坚持做一件长期主义下的突破性尝试:探索不可成药靶点,探索“无药可治”新解法,探索一条难而险的新兴技术路径,而核心,始终是受急迫的需求使然。
ReviR的目标,不止是成为一家驱动药研、解码“不可成药”靶点的平台型公司,更要做独立研发。目前,ReviR管线研发聚焦的三大重点领域各有考量:肿瘤领域大而广,仍有许多待挖潜赛道与机制;神经系统领域,极度缺乏与肿瘤药物研发类似的项目积累与沉淀,也还需提升临床试验效率;罕见病,患者基数不容小觑,不可成药靶点较多,但生物技术企业研投意愿不足。
尤其是在中国罕见病药品问题上,本质是创新药行业面临的所有问题的集合。罕见病致残率高而可治性低,但有明确治疗方案的仅约5%,亟待破局。同时,罕见病往往因病因繁多、症状复杂,并且同种疾病之间可能会有不同症状,患者容易遭到误诊、漏诊。打通罕见病药物可及性,补齐罕见病治疗领域的“缺角”,是前沿人工智能方法和新兴生物技术存在最为关键的意义之一。
而在罕见病上,从亨廷顿舞蹈症、渐冻症,再到腓骨肌萎缩症,ReviR的选择,一直都是在临床空白的领域寻求颠覆性突破,将不可能努力逆转成为一种可能。本次在CMT领域的探索和研发投入是一个重要的节点,CSO Paul August表示,在未来,ReviR的目标是不断投入研发这种可口服的基因疗法,最终将其扩展到更广泛的疾病领域。
一审| 黄佳
二审| 李芳晨
三审| 李静芝
基因疗法临床研究临床结果
2024-06-05
关注并星标CPHI制药在线
一、腓骨肌萎缩症简介
腓骨肌萎缩症(Charcot- Marie- Tooth diseases,CMT)是一种常见的遗传性周围神经疾病。在1886年由法国Charcot、Marie和英国Tooth三名医生报道,因此用三位医生名字首字母命名CMT。“CMT”和“渐冻症”不能混为一谈,是有本质区别的,CMT是遗传性运动感觉神经病,主要侵犯周围神经。而“渐冻症”是一个通俗的叫法,这个疾病的专业名字叫运动神经元病,有多个类型,最常见的类型是肌萎缩侧索硬化症(渐冻症),跟老年痴呆、帕金森病一样,属于中枢神经系统退行性疾病。腓骨肌萎缩症临床特点包括对称性远端为主的肌无力伴萎缩、感觉减退以及弓形足、脊柱侧凸等骨骼畸形。临床上根据神经电生理、病理学和遗传学特点可分为多种亚型,基因检测手段有助于明确其致病基因。康复治疗、外科矫形手术治疗和改善症状的药物治疗有助于缓解疾病症状,减轻骨骼畸形。部分针对病因和发病机制的特异性治疗药物已进入临床试验阶段,其疗效和安全性有待进一步明确。
CMT的患病率约为40/100000,是2018 年国家卫生健康委员会发布121种《第一批罕见病目录》中的其中一种疾病,但却是遗传性周围神经病中最常见的一种。临床特点包括:儿童或青少年起病,慢性进行性的对称性肌无力及肌萎缩、远端型感觉障碍、腱反射减弱或消失、弓形足等骨骼畸形。CMT具有显著的遗传异质性和临床异质性,相同的基因突变可导致不同的临床表型,目前已有大约100个不同的CMT致病基因相继被克隆并报道。
CMT的发病过程表现各异,其中多数患者在出生时无明显异常,但随着生长发育,逐渐出现足踝部力量减弱或步态改变等轻微症状。当家长发现并带患儿前往当地医院就诊时,有很大一部分情况下医生难以立即确定病因。随着时间的推移和疾病的逐步进展,患者的足踝畸形会进一步加剧,表现为踮脚、内翻、乏力以及下蹲困难等不同程度的身体在病情严重的情况下,患者可能需要依赖拐杖行走甚至乘坐轮椅。
CMT的发作时间点存在较大的差异,统计显示,约有50%的患者在10岁前即表现出相关症状,而70%的患者则在20岁前发病。其余患者则可能在20至30岁,甚至更晚的年龄段才出现病征。因此,在发病早期的患者群体中,主要为青少年患者。
二、遗传方式及临床分型
根据神经电生理和神经病理学的特征,临床上将CMT细分为以下三种类型:
(1)脱髓鞘型 CMT:其显著特点在于神经传导速度(NCV)明显减缓,通常上肢运动的NCV低于38 m/s。进一步通过神经活组织检查(活检)结果显示出显著的周围神经髓鞘异常,由此特征区分此类型CMT。
(2)轴索型 CMT:其特点在于周围神经NCV略有减缓或基本接近正常值,上肢运动的NCV一般超过38 m/s。神经活检的病理结果则提示存在慢性轴索变性及再生现象,这是该类型CMT的主要诊断依据。
(3)中间型 CMT:该类型介于前两者之间,上肢运动的NCV介于25~45m/s之间。
此外,CMT的遗传方式多样,以常染色体显性遗传为主,但也可能表现为常染色体隐性遗传或X连锁显性或隐性遗传。结合这些遗传方式、临床表现、电生理和病理特点,我们可以进一步将CMT细分为多种亚型,以便更精确地诊断和治疗。
三、临床表现
该病显著特点是对称性、缓慢进行性的四肢周围神经脱髓鞘和轴索变性 , 造成肢体远端肌肉的萎缩和无力,进而出现肢体变形。典型表现有:
(1)高弓足:CMT第一个特征就是足弓很高,逐渐发展为弓形足。
(2)跨阈步态:一般早期是无意中发现脚踝力量减退,随着病情的发展,双小腿缓慢进行性萎缩及无力,呈“仙鹤腿”或“倒酒瓶”样改变。同时伴有行走及平衡障碍,为避免摔倒,患者会将膝部异常高抬而表现出“跨阈步态”。部分患者的大腿肌肉也会出现无力。
(3)腕部、手部肌肉萎缩:出现手部肌萎缩,难以完成一些精细运动,如书写、从地面拾起细小物品等。后期可以出现抓握困难,部分患者累计上臂。
(4)感觉障碍:不是腓骨肌萎缩症的突出表现,患者可以有末梢型感觉减退和麻木,呈现手套袜套样感觉障碍,部分患者可以合并疼痛。
(5)其他:有些患者可以出现听力减退,视神经萎缩和脊柱侧弯等骨骼发育异常。
四、检查
1、神经电生理检查
神经电生理检测,特别是NCV检测,对于明确周围神经脱髓鞘或轴索损害具有显著意义,对于CMT的分型也当检测到NCV的匀一性减慢,即上肢正中神经或尺神经的运动传导速度低于38 m/s时,这提示我们可能面临脱髓鞘型的CMT,例如CMT1和CMT4。
然而,当NCV仅出现轻度的减慢或保持正常状态(即上肢正中神经或尺神经的运动传导速度超过38 m/s),同时伴随复合肌肉动作电位或感觉动作电位波幅的降低,这是CMT2的经典表现。在某些中间型情况下,即上肢运动的NCV介于25~45 m/s之间,特别是针对男性患者,我们还应警惕可能存在CMTX的风险。此外,不论患者性别如何,显性遗传的中间型CMT同样是一个可能的诊断方向,尽管这种情况相比CMTX更为少见。
在肌电图检查方面,常见的特征是运动单位电位的时程较长、波幅较高,同时募集相减少,而纤颤电位相对罕见,这些表现都提示了慢性失神经支配和神经再生的现象。
此外,NCV检查还是区分CMT与其他获得性多发性周围神经病的重要手段。在CMT1中,NCV的减慢通常十分显著且基本对称,运动神经很少出现传导阻滞或动作电位波形离散。相比之下,获得性脱髓鞘性周围神经病如CIDP的NCV减慢并不那么显著,并常呈不对称性,伴有运动神经传导阻滞或动作电位波形离散的特点。然而,值得注意的是,CMTX1型患者可能会有不对称性的NCV减慢,甚至伴有波形离散或传导阻滞的情况。更为罕见的是,由MPZ基因突变导致的CMT1B也可能出现传导阻滞。电生理检测不仅有助于鉴别CMT,还能帮助我们区分其他神经肌肉疾病如远端型肌病/肌营养不良、dHMN等
2、基因检测
基因检测在确诊CMT的过程中扮演着至关重要的角色。随着二代测序技术的广泛应用,包括疾病特异性基因检测、全外显子测序、全基因组测序以及高通量转录组测序等,我们已经能够发现越来越多的CMT致病基因及其突变位点。其中,PMP22、MPZ、GJB1、MFN2和GDAP1等基因被认为是CMT的主要致病基因。
在针对常染色体显性遗传的CMT1和散发CMT1患者的诊断中,我们通常会首先采用多重连接探针扩增技术(MLPA)来检测PMP22基因的大片段重复突变。如果检测结果为阴性,并且在家系内没有出现男性传男性的情况,那么我们会考虑CMTX的可能性,并进一步对GJB1基因进行突变分析。如果排除了CMTX1的可能性,我们还将对PMP22和MPZ基因进行点突变检测。
对于疑似CMT2的患者,我们会首先进行MFN2和MPZ基因的突变检测。对于家系内无男传男现象的病例,尤其是女性CMT2患者,我们同样会考虑CMTX的可能性,并对GJB1基因进行突变分析。
对于中间型NCV的患者,我们会优先进行GJB1、MPZ、NFL、GDAP1等基因的突变分析。而对于常染色体隐性遗传的CMT患者,无论是脱髓鞘型(CMT4)还是轴索型CMT2,我们都将首先进行GDAP1基因检测。
如果在一个疑似CMT的家系中,基因检测结果为阴性,那么我们可能需要考虑是否可能存在其他类型的遗传性周围神经病,例如dHMN、遗传性感觉自主神经病等。在这种情况下,我们可以进一步进行相关基因检测。同时,也不能排除存在未报道的新CMT致病基因的可能性。为此,我们可以采用全外显子测序或全基因组测序的方法来寻找可能的新的致病基因突变,并进行功能验证。
3、神经活检病理
基因检测技术的日益普及,使得神经活检病理检查在多数CMT(遗传性运动感觉神经病)病例的诊断中变得不再必须。不过,对于那些诊断颇具挑战的散发型病例,或者基因检测结果为阴性的家族性病例来说,周围神经活检的病理检查结果仍能给予关键的诊断线索。
通过光镜观察,我们可以看到周围神经在经过甲苯胺蓝染色后展现出有髓纤维的多样化大小。而在电镜的放大下,薄鞘纤维和裸轴索纤维的形态清晰可见。当施万细胞增生形成特殊的“洋葱头”状结构时,这往往暗示着CMT1类型的CMT。另一方面,有髓与无髓纤维的减少以及再生簇的形成,则倾向于表明CMT2类型的病情。
在CMTX的病理表现上,我们可以看到轴索的丢失以及部分脱髓鞘的改变并存,但“洋葱头”结构相对较少值得注意的是,某些独特的髓鞘病理改变与特定的基因变异相关联。举例来说,MPZ基因变异的患者中,常常可以看到髓鞘的松解与肿胀;而在MTMR2基因或FGD4基因突变所导致的CMT4患者中,可以观察到大量髓鞘向外折叠的病理特征;至于NEFL基因突变的病例,则常呈现巨轴索的现象。这些病理改变不仅有助于我们更准确地诊断病情,也为后续的治疗提供了宝贵的参考依据。
五、治疗
目前尚无有效逆转CMT病程的治疗方法,支持性治疗包括康复治疗、外科矫形手术治疗和缓解症状为主的药物治疗等,同时需要多学科团队相互协作。已有研究针对不同CMT亚型的发病机制和治疗靶点研发了相应的特异性治疗药物,但绝大多数处于药物临床试验阶段,尚未进入临床治疗。
1、康复治疗
多种康复治疗手段已被用于CMT的治疗。有研究结果表明,轻到中等强度的运动训练能有助于CMT患者改善下肢肌力及行走能力。适当的有氧训练能够改善患者的体能,提高有氧运动功能。下肢的被动拉伸训练有助于预防和改善跟腱挛缩。适当穿戴辅助器具如矫形器能够改善患者的姿势和平衡功能。个体化的踝?足矫形器有助于改善足下垂,辅助患者行走并缓解足弓疼痛。
2、外科矫形手术治疗
多数患者在儿童和青少年时期足部的内翻畸形是柔软可恢复的,此时应首先选择非手术治疗。随着病情的发展,足内翻逐步进展加重为固定畸形,无法穿戴矫形器具,此时需要采取外科手术治疗。手术治疗的原则是纠正足部畸形,重建和平衡足踝肌力。手术方案包括肌力平衡手术(各种类型的肌腱转移术)、软组织松解与矫形(足底筋膜切开术)、跟骨截骨矫形、腓肠肌复合体处理、踇趾矫形或其他足趾矫形手术。
3、缓解症状的药物治疗
CMT患者饱受疼痛之苦,该症状在23%至85%的病患群体中都有所体现。这种疼痛常与骨骼形变、长时间姿势不当或是肌肉疲劳紧密相连。为了缓解这一症状,除了常规的物理康复、佩戴矫形器械和进行外科矫形手术外,非甾体类抗炎药如对乙酰氨基酚也是有效的治疗手段。
对于神经性疼痛尤为明显的患者来说,药物选择需更为细致。首选的是那些针对神经病理性疼痛的特效药,如三环类抗抑郁药、选择性五羟色胺再摄取抑制剂以及抗惊厥药(普瑞巴林、加巴喷丁、卡马西平等)等。同时,应尽量避免使用阿片类药物,以免给患者带来更多负担。
此外,CMT患者还普遍感到易疲劳,这可能与肌力减退及心肺功能下降有关,部分患者还可能与阻塞性睡眠呼吸暂停有关。根据研究,莫达非尼在一定程度上能够改善这一情况,但使用该药物时需注意可能产生的不良反应。
最后,需特别强调,患者应避免使用可能加重周围神经损伤的药物,包括但不限于长春新碱、铂类、紫杉醇衍生物、硼替佐米、呋喃妥因、氨苯砜、来氟米特、甲硝唑、司他夫定、他克莫司等。这些药物的潜在风险不容忽视,患者应遵循医生的建议,合理选用药物。
4、特异性药物治疗
对于CMT发病机制的相关靶点,当前的研究重点主要侧重于PMP22基因突变所引发的CMT1A。CMT1X是一种表现为进行性肌肉萎缩、四肢无力和感觉丧失特征的遗传性神经病变。它主要由连接蛋白32(Cx32)的突变造成,这种蛋白是形成髓鞘间隙连接通道的重要成分,对神经功能和完整性具有关键作用。
1)维生素C
在体外细胞实验中,维生素C被发现有助于促进髓鞘形成。在动物实验层面上,高剂量的维生素C被证实能够改善CMT1A模型鼠的运动表现。然而,在临床实践中,尽管尝试了不同剂量的维生素C(1~4 g/d)治疗,但并未能显著改善CMT1A患者的临床症状。因此,维生素C在CMT1A治疗中的实际效果仍需通过更多的临床实验来进一步探索。
2)黄体酮及其衍生物
动物实验结果还显示,黄体酮及其衍生物可能会加剧CMT1A大鼠的神经功能缺损和神经病理损害。但值得注意的是,奥司那酮,一种黄体酮拮抗剂,已被证实在降低CMT1A大鼠的神经周围PMP22蛋白表达及改善疾病表型方面具有积极效果。遗憾的是,由于奥司那酮的毒副作用较大,使其在临床治疗中的应用受到严重限制。不过,ulapristal,作为奥司那酮的生物等效剂,展现出了较好的安全性,这可能为未来的CMT1A治疗提供了一种新的选择。
3)神经营养因子3(neurotrophin-3,NT3)
在法国开展的 ulapristal治疗CMT1A 的Ⅱ期临床研究正在进行中。神经营养因子3(neurotrophin-3,NT3)能够促进轴突生长,在一项随机、双盲、安慰剂对照试验中,8例 CMT1A患者接受了6个月的皮内NT3注射治疗, NT3似乎能够改善患者的感觉神经受累程度,增加腓肠神经小直径有髓纤维的数量。
4)PXT3003
PXT3003作为复方药剂,主要包含小剂量巴氯芬、山梨醇及纳曲酮等成分。根据临床前期研究,此药能有效减少PMP22转基因大鼠体内PMP22蛋白表达,并优化髓鞘的生成过程。此前,PXT3003已在法国进行了多中心Ⅱ期临床实验,并完成了欧美多中心Ⅲ期临床研究。这些研究结果显示,相较于安慰剂组,高剂量组的CMT1A患者在若干关键指标上均取得了显著的进步,同时该药物的安全性也得到了验证。不过,在Ⅲ期临床研究中,高剂量组制剂出现了析晶问题,这导致约一半的患者被迫提前退出试验。因此,美国食品药品监督管理局要求在欧洲和美国再次进行一项Ⅲ期临床试验,以进一步验证PXT3003的治疗效果。与此同时,国内也正积极开展关于PXT3003治疗CMT1A的多中心、随机、双盲、安慰剂对照Ⅲ
为了进一步研发出既安全又高效的治疗方案,本研究正致力于设计一种新型适配体偶联纳米颗粒,这种纳米颗粒能够表达Cx32基因,并特异性地作用于CMT1X小鼠模型的雪旺细胞。我们相信,通过这种方法,可以实现更加精准的生物分布,并可能为CMT1X等由雪旺细胞基因缺陷导致的脱髓鞘神经性疾病提供更安全、更具可操作性的基因治疗手段。
5)新型适配体偶联纳米颗粒
2023年9月12日,肌萎缩症协会(MDA)和Charcot- Marie - Tooth协会(CMTA)宣布了一项总额为299,992美元的合作研究资助,用于测试使用纳米颗粒向雪旺细胞传递基因以治疗CMT1X。这项为期三年的研究名为“基于纳米颗粒的施旺细胞基因递送治疗CMT病”,将由塞浦路斯神经病学与遗传学研究所的Alexia Kagiava医学博士领导。这项研究的积极结果将为周围神经系统基因传递提供一种新的策略,预计比腺相关病毒(AAV)基因传递策略更有针对性,更安全,可用于临床转化。新的雪旺细胞靶向纳米颗粒递送也有望应用于其他形式的Charcot- Marie - Tooth(CMT)和其他由雪旺细胞基因缺陷引起的脱髓鞘神经病变。为了开发一种更安全、更有针对性的方法,本研究旨在设计一种携带表达Cx32基因的新型适配体偶联纳米颗粒,使基因能够特异性地进入CMT1X小鼠模型中的雪旺细胞。这种靶向纳米颗粒方法有望实现更有针对性的生物分布,并为CMT1X以及其他由雪旺细胞基因缺陷引起的脱髓鞘神经性疾病提供更安全、更可转化的基因治疗
6)CKD-510
韩国经济日报2023年11月6日消息,韩国制药公司钟根堂(CKD制药集团)的股价在周一飙升逾25%,原因是该公司与诺华医药(Novartis)签署了一项价值13亿美元用于治疗腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)的外包许可协议,这是该公司有史以来最大的一笔交易。
该公司周一宣布,它与诺华签署了13亿美元的授权协议以授予这一瑞士跨国制药公司开发并商业化其所研发的新药CKD-510的权利,CKD-510是专门为罕见病CMT研究的药物,根据协议,诺华将向钟根堂支付8000万美元的预付款,并根据后期药物的开发、获批情况再支付12亿美元,另外还有单独的销售版税收益。这是这家韩国制药公司成立80多年以来最大的对外授权交易,也是整个韩国制药行业今年最大的交易。
CKD-510是由钟根堂制药开发的一种作用于组蛋白去乙酰化酶6(HDAC6)的小分子抑制剂,可用于调节各类炎症疾病。韩国制药商钟根堂在临床前试验中证明了CKD-510在治疗心血管疾病和其他hdac6相关疾病方面的疗效,并在法国的1期临床试验中证明了其安全性和耐受性.
众所周知,CMT会损害周围神经,由于手脚肌肉萎缩、运动和感觉功能的丧失,病人难以行走并丧失活动能力。虽然目前还没有完全授权的治疗该遗传性疾病的方法,但美国食品和药物管理局(FDA)在2020年3月认定了CKD-510治疗CMT的孤儿药资格。
六、小结
CMT 是最常见的周围神经遗传性疾病,具有显著的临床表型和遗传表型异质性。随着二代测序等基因检测技术的发展和普及,越来越多的 CMT致病基因和新发突变位点被发现和克隆,极大地丰富了 CMT的基因谱。康复训练治疗和外科矫形手术治疗仍是 CMT的主要治疗手段,针对发病机制中重要靶点的多种特异性治疗药物的研究处于临床试验阶段或临床前研究阶段,有望将来为CMT的治疗带来新的突破
参考文献
1.腓骨肌萎缩症的诊治,姚晓黎, 何若洁. 中华神经科杂志, 2024,57 (03)290-297.DOI:10.3760/cma.j.cn113694-20230915-00165
2.Https://www.kedglobal.com/bio-pharma/newsView/ked202311060009
【企业推荐】
【智药研习社直播预告】
来源:CPHI制药在线
声明:本文仅代表作者观点,并不代表制药在线立场。本网站内容仅出于传递更多信息之目的。如需转载,请务必注明文章来源和作者。
投稿邮箱:Kelly.Xiao@imsinoexpo.com
▼更多制药资讯,请关注CPHI制药在线▼
点击阅读原文,进入智药研习社~
临床研究基因疗法
2023-07-19
·药时代
正文共:24954字 38图预计阅读时间:63分钟药时代编者按《药物递送——生物医药未来的“卡脖子”技术》由谢雨礼博士倾情撰写,全文2万多字,全方面描绘了药物递送技术这一近年来备受关注的火热领域,业界流传“得递送者得天下!”这样一句话。本文虽无华美的辞藻,但笔翰如流未尝壅滞,文采斐然,字字珠玑。通篇读下来,酣畅淋漓,只感茅塞顿开、大受裨益。作为药时代专栏作家,谢雨礼博士在药时代平台发表了诸多爆款好文。欢迎大家在“谢雨礼博士专栏”中查阅其它精品文章!前言递送技术被认为是药物开发的最后一公里,其重要性无需赘述。人们早就注意到了口服、吸入和外敷等给药方式和制剂对药物疗效和安全性的重要性。普通的药物制剂进入人体后立即释放,药物的药代动力学(DMPK)更多地依赖于药物内在的理化性质。通过制剂或者装置控制药物释放的递送系统起步于上个世纪50年代,标志性产品是1952年上市的治疗多动症的Spansule®缓释胶囊。该技术通过高分子包衣的溶出速度来控制药物右旋苯丙胺的释放速度,实现了12小时缓释的效果。自此,药物递送技术伴随着现代生物医药走过了70年的发展历程,取得了令人叹为观止的进步,其中递送mRNA疫苗的脂质纳米颗粒(LNP)成为人类这次抗击新冠的关键技术之一(图1)。图1:药物递送技术的70年发展历程(1952-2022)药物递送的挑战现代生物医药以1953年DNA双螺旋结构的发现为起点,沿着生命“中心法则”由外及里全面覆盖了蛋白质、核酸和基因三个环节,包括最早靶向蛋白质的传统药物、重新崛起的核酸药物以及最新的精确操控RNA和DNA分子的基因疗法和基因编辑等。药物形式(modality)除了经典的小分子,多肽、蛋白、多糖和核酸,还出现了细菌、细胞和病毒等更加复杂的活体药物。读者也许注意到了,现代生物医药和递送技术的起步前后相隔不到一年,发展几乎同步。这也不难理解,未满足临床需求推动新技术的发展,而新技术对药物递送不断提出新课题。如表1所示,每种药物形式或者技术面临的递送挑战不尽相同。表1:各种药物形式或者技术面临的递送挑战药物递送的两个目的当前,开发给药系统和递送技术的出发点分为两种。对于化药和单抗等相对成熟的药物,目标主要是改善现有药物的缺点,也就是我们常说的改良新药。在新实体分子的开发中,制药企业为了追求速度,制剂或者给药系统往往不是最优的,这给改良新药带来很大的空间(表2)。值得注意的是,改良新药即使只是改善病人的服药依从性,临床意义也不容忽视。据报道,慢性病人不按说明书服药的比例达到50%,仅美国每年因为服药的不规范导致12万以上的死亡,10%的住院以及超过1000亿美元的额外医疗费用(1)。表2: 改良新药的作用和商业化实例递送技术的第二个目的就是赋能新技术。对于PROTAC、核酸药物、mRNA疫苗、基因编辑和细胞疗法等领域,优化药物本身和普通给药系统很难解决成药性的问题,递送技术成为这些新技术不可分割的一部分。核酸药物的潜力早在20年前就广为人知,但直到GalNac偶联技术和LNP给药系统的成功,才迎来了快速的发展,但仍很大程度上局限于肝病或肝靶点介导的疾病。基因编辑等新技术更是受到递送技术的制约而不能完全释放其治疗潜力。发明基因编辑工具的顶尖科学家张锋和刘如谦近年来将很大精力放在了开发新的递送系统,最近发表的包括全新的类病毒颗粒(VLP)和细菌蛋白注射器(PVC-eCIS)等(2,3,4)。递送技术还可以将活体药物的个性化生产转化为常规的批生产。CAR-T等个性化细胞疗法被认为是肿瘤免疫疗法的革命性技术,但急需解决成本高、价格昂贵的问题。最近,欧美开始尝试通过靶向递送实现在病人体内直接改造免疫细胞,有望解决当前细胞治疗个性化生产、成本高的痛点。解决药物递送的三种方案药物递送无非就是要将适量的药物递送到人体或某个特定部位并保证合理的药物浓度和作用时间。如表3所示,要实现或者提升药物递送效果,宏观上有三种方案(5)。表3:解决药物递送问题的三种方案 第一,通过改造或者化学修饰优化药物分子本身,提高成药性并使得药物可以方便地通过常见的方式和成熟的制剂技术给药。对于常规药物,这本身就是药物化学或者蛋白工程的一个工作内容。药物分子优化得越好,后续制剂开发或者给药的难度越小。除了结构改造和晶盐型筛选,小分子药物研发中常见的氘代和前药技术实际上也是一种解决药物递送的策略,最近例子包括上海药物所和君实生物联合研发的口服抗新冠药物氢溴酸氘瑞米德韦片(vv116)。对于多肽和蛋白药物,使用非天然氨基酸和化学修饰(如PEGylation)是提高药物稳定性、延长半衰期的常见办法。49个批准的GPCR多肽药物中21个同时具有化学修饰和非天然氨基酸,比如治疗中枢性尿崩症的去氨加压素(Desmoppressin)。PEG修饰的多肽还包括最近火爆降糖和减肥市场的GLP-1药物。1990年获批的Adagen(pegademase bovine)是一种PEG修饰的蛋白药物、来源于牛的腺苷脱氨酶用于治疗伴有腺苷脱氨酶缺乏的免疫缺陷疾病。2008年上市的抗炎药物赛妥珠Cimzia是一款PEG修饰的抗TNF单抗。PEG结构上与水分子有类似之处,水溶性好且不易被降解或者免疫清除,是理想的化学修饰工具。除了多肽和蛋白外,PEG也用于小分子和载药系统的修饰,如PEG化纳洛酮Movantik®,阿霉素PEG化脂质体Doxil®和Patisiran-PEG化纳米颗粒 Onpattro®等。Extend公司通过化学修饰开发了一种多肽和蛋白药物皮下给药的技术(D-VITylation®)。其原理是用维生素D的类似物修饰多肽和蛋白,皮下注射后药物被维生素D结合蛋白(VDBP)识别并转运至血液循环系统中,从而改善生物利用度并延长多肽和蛋白药物的半衰期。核酸药物的潜力巨大,但递送的难度却比传统药物大得多,主要原因包括:1)核酸的分子量和负电荷让其不能自由通过生物膜;2)RNA容易被血浆和组织中RNase酶降解,被肝脏和肾脏快速清除,以及被免疫系统识别清除;3)进入细胞后“卡”在内吞小体中无法发挥功能。要解决这些问题,除了利用药物递送系统,化学改造必不可少。改造的目标是让其稳定、躲避免疫系统的识别并保持生物功能。如图2所示,核酸的化学改造按部位分三类:碱基,糖环和连接基团磷酸的改造(6)。这两年,Orna 、Laronde和环码生物等新锐公司开拓的不需要添加帽子和尾巴结构的环形RNA技术可以算作更加激进的化学改造。图2:核酸药物中常见的核苷酸化学改造化学改造的重要性不言而喻。BioNTech副总裁、时任宾夕法尼亚大学研究人员的Katalin Karikó等人发现核酸分子通过化学修饰能够逃逸机体的免疫防御。使用化学修饰的N1-甲基-假尿嘧啶是这次mRNA新冠疫苗成功的关键因素,Katalin Karikó作为主要贡献者成为诺奖呼声很高的人选。DS-8201在乳腺癌治疗上的突破让ADC成为行业热点,也是中国Biotech与跨国公司合作交易最活跃的领域。ADC为代表的偶联药物实际上是一种通过化学改造赋予药物本身靶向性的递送技术。ADC最初的设计思路就是利用单抗将化疗毒素靶向递送到肿瘤组织从而减少对正常组织的杀伤。当然,不同于常规的药物递送系统,ADC是将小分子和单抗通过连接子组合的单一药物形式。组合往往带来不同于基本单元的特点和性质。当前ADC药物的设计理念已经超出了递送的范畴。比如DS-8201所用的Her2单抗除了肿瘤靶向功能,本身也有治疗效果,甚至能够与所连接的毒素Dxd形成协同作用。类似地,多肽偶联药物PDC、小分子偶联药物SMDC,核素偶联药物RDC以及小核酸的GalNac偶联都可以看作一种化学修饰的递送技术。其中,GalNac被证明能够高效干净地将核酸药物带到肝脏组织,是推动核酸药物第二次浪潮的关键技术。CAR-T,CAR-TCR,CAR-NK,CAR-M, TILs,TCR-T等细胞技术将免疫细胞通过基因改造,装上识别肿瘤细胞的单抗,在某种意义上也是通过改造药物解决药物递送的策略。同理,基因工程改造的微生物和病毒,如新一代的溶瘤病毒序列中插入PD-1单抗,炎症因子等,既是药物组合的平台,也是一种递送工具。 第二,通过改变人体吸收或者药物作用的微环境提升药物的递送效果。我们所服用的药片或胶囊不只是包含药物成分,还包括多种辅料和添加剂。这些辅助成分的作用就是改变药物吸收的环境比如胃肠道的酸碱性和细胞膜结构,从而影响药物的吸收,代谢和分布。药学中的处方研究其实就是筛选辅料和添加剂的种类和比例,找到最有利于药物递送的一种组合。因为药物本身的局限性,有时常规处方不能解决问题,还需添加特别的成分,甚至包括辅助药物。辉瑞公司推出的抗新冠小分子药物PAXLOVID实际上包含两种药物成分,其中只有化合物PF-07321332具有抑制新冠病毒的功能,另外一个成分Ritonavir是代谢酶P450的抑制剂,所起的作用是减少PF-07321332的肝代谢,增加主药的口服生物利用度。制剂技术的一个难点是实现多肽和蛋白药物的口服给药。Emisphere开发的吸收增强剂SNAC在胃内使得胃黏膜表面PH值短期局部升高,从而使得胃蛋白酶失去活性阻止药物降解,同时胃局部亲脂性增加,使多肽和蛋白更易通过细胞膜进入血液。使用这项技术,诺和诺德在2019年推出了一款口服的GLP-1多肽药物Rybelsus(索马鲁肽)。当然,这款产品的口服生物利用度仍然比较低,还有很大的改进空间。另外,口服的小分子GLP-1药物也已陆续进入临床研究,有望进一步优化这类重磅减肥药的依从性问题。i2o Therapeutics开发的离子液体(ionic liquid)技术宣称也可用于口服递送多肽、蛋白等生物制剂,但未见临床产品报道。值得注意的是,Oramed添加代谢酶抑制剂的口服胰岛素未能达到预期效果,已经宣布终止针对糖尿病的三期临床研究。单抗药物一般需要住院静脉注射,给病人带来诸多不便。皮下或肌注给药则可以通过预填充注射器居家给药,不但改善病人的依从性还能有效地降低医疗成本。最近,透明质酸酶成为单抗皮下给药的新技术(ENHANZE®),其原理是利用透明质酸酶降解皮下的透明质酸,清除大分子皮下渗透的障碍,实现单抗的皮下高剂量快速给药(图3)。2019年FDA批准了用于皮下注射的曲妥珠单抗和透明质酸酶-ysys注射液(Herceptin Hylecta),给药时间从3小时缩短至5分钟。2020年FDA还批准了基因泰克的两个Her2单抗和透明质酸酶的三药组合Phesgo,以及强生的CD38抗体Darzalex皮下注射剂Darzalex Faspro。当然,单抗的皮下给药也可以通过第一种策略即优化药物实现,比如使用比较小的纳米单抗,具体例子包括康宁杰瑞开发的全球首款皮下给药PD-L1单抗。图3:透明质酸酶技术的工作原理CAR-T输注前的清淋预处理实际上也是一种改变微环境的递送策略。通过化疗或放疗,清除患者体内的淋巴细胞。清淋预处理消除调节性T细胞为CAR-T细胞在体内的扩增和存活创造了一个“有利”的环境。此外,还可以上调肿瘤免疫原性并改善疾病控制。 第三,开发药物递送系统和装置,在药物和体内环境之间添加一道屏障,实现药物的控释。药物的改造和处方开发一般归属于药物发现和药学研究的一部分。多数情况下,递送技术专指药物的递送系统和装置。如图4所示,各种药物递送系统或者装置按给药途径又分为口服、透皮、埋入、吸入和注射等几大类型(5)。除了少数需要机械控制外,基本原理都是用脂类、蛋白质和病毒等天然材料,或者高分子,无机盐和硅胶等生物相容性材料包裹药物,并利用这些材料的溶解、扩散、渗透、离子交换、降解和化学反应来控制药物的释放。图4:常见的药物递送系统和装置早期小分子化学药物在市场上占据统治地位,改良制剂主导了药物递送的发展,代表性技术包括透皮贴剂、口服缓释制剂、埋入装置、复杂注射剂(脂质体、乳剂、微球、纳米颗粒和纳米混悬剂)和吸入剂(气雾剂,干粉剂和喷雾剂)等。传奇人物Alejandro Zaffaroni博士创立的ALZA是制剂技术公司中的佼佼者,开拓和发展了透皮制剂和渗透泵(OROS)等技术,后被强生收购。以ALZA为代表的制剂公司开发了多款透皮制剂药物,年销售超1亿美元的产品有12个,其中芬太尼透皮贴剂Duragesic的年销售峰值超过30亿美元。今天,透皮给药系统发展为三种类型,除了扩散型的贴剂外,还包括离子电渗给药(Zecuity舒马曲坦)和微针阵列贴片( Fluzone流感疫苗)。渗透泵技术利用高分子填充层吸水后膨胀将药物从带有单室、多室或者多个膜孔的片剂和胶囊中推出,可以实现药物的零级动力学释放,从而更好地维持药物的血药浓度。ALZA推出的渗透泵产品Procardia XL(硝苯地平,高血压)和Concerta(盐酸哌甲酯,多动症)相继成为年销售额超过10亿美元的重磅产品。口服缓控释系统还包括将药物和亲水凝胶、不溶性、溶蚀性惰性材料,或者离子树脂制成骨架型或微丸片剂、胶囊、口服溶液、混悬液等缓控释制剂。亲水凝胶吸水后膨胀,形成的凝胶屏障可减慢药物释放速率。不溶型骨架片的骨架孔隙渗满消化液时,药物发生溶解并缓慢扩散。溶蚀骨架被溶蚀后生成蜡类物质或惰性脂肪,通过化学蚀解或孔道扩散达到缓释的作用。而载药的树脂进入胃肠道后发生离子交换而慢慢释放药物。微丸片剂是一种更加复杂的多单位缓释系统,一片药物包含数以百计微丸亚单位,服药后广泛分布于整个胃肠道。单个微丸的质量偏差对片剂的整体释放情况影响极微,显著提升了药物释放的均一性和效率,具体例子包括治疗高血压的美托洛尔微丸缓释片等。1989年武田与雅培联合开发的亮丙瑞林PLGA微球注射剂(Lurpo Depot®)为长效注射剂和植入装置打开了大门,将药物递送的时间从几天延长到数周,甚至几个月。与此同时,长效或者复杂注射技术成功地推广到多肽和蛋白药物的给药。代表性的复杂注射剂除了微球,还包括脂质体(阿霉素Doxil®),纳米颗粒(紫杉醇-白蛋白复合物Abraxane®),纳米晶体(西罗莫司纳米晶制剂Rapamune®),和脂肪乳(阿瑞匹坦Cinvanti®)等。植入剂在缓释载体之外还加有控制药物扩散的装置,可以进一步延长药物的释放周期。如避孕埋入装置Norplant,是一根火柴棒大小的圆柱形硅橡胶管,其药物释放周期长达5年。Taris公司硅酮埋入装置是基于MIT教授Robert Langer 和Michael Cima的研究成果开发的一种泌尿系统递送技术,可使用微创方法植入膀胱。TARIS系统可以在膀胱中以相对较高的浓度长期放置化疗药物和靶向治疗,效果优于口服或注射药物。吸入给药直接靶向病灶或者通过鼻粘膜入脑,特别适合激素等高活药物的递送,在呼吸和神经系统药物中得到广泛应用。目前,已有60多种吸入产品上市,包括多个治疗哮喘和慢性阻塞性肺疾病(COPD)的重磅药物。吸入疗法除了对制剂有较高的要求外,还必须借助雾化吸入器(nebulizer,NEB)、定量吸入器(pressured metered-dose inhaler,pMDI)和干粉吸入器(dry powder inhaler,DPI)等装置进行精确和有效的递送。雾化吸入器笨重不易携带,应用受到很大限制。自1956年的Medihaler问世以来,定量吸入器成为使用最为广泛的吸入器,80%以上的哮喘患者均采用此法。干粉吸入器是呼吸触动型,药物粉雾的产生通过患者的吸气产生,不需要协调配合,容易为儿童或老人所接受。特别是1988年推出的多剂量干粉装置Turbuhaler,含有200个剂量单元,患者吸入前只需旋转吸入器底部便可释放出一个剂量,它的出现对其他吸入装置的有效性和方便性形成了真正的挑战。市场上,干粉吸入剂产品迅速上升,占比达到55%,显示出巨大的商业前景。除了上述常见的给药系统外,还有多种针对不同目的,用途和人群的创新制剂技术,比如舌下片,口腔膜,栓剂,各种儿童剂型和妇科产品等。药品的制剂创新没有止境,是一个持续的过程。比如,非载体抗炎药物双氯芬酸自1973年第一次上市,几十年间共推出17款不同的制剂产品。新型递送材料,装置以及人工智能和大数据在处方设计中的应用必将推动给药系统和制剂技术的进一步发展。值得一提的是,高端给药系统或者制剂的生产工艺复杂,即使专利到期也很难仿制。比如PLGA微球制剂上市已超过三十年,但市场上尚无FDA批准的长效微球仿制产品。造成这种情况的原因之一是原研公司没有披露详细的PLGA表征数据和工艺参数。吸入产品涉及制剂和装置的配合,面临同样的困境。这也是高端制剂不同于新实体分子的一个特点,没有专利悬崖的威胁,源源不断地给原研公司带来巨大利益。比如,亮丙瑞林微球注射剂从1992年开始销售额始终保持在10-20亿美元,是武田崛起为跨国大公司最重要的支撑之一。政府从降费的角度也注意到了这个问题,前FDA局长Scott Gottlieb就曾提出要降低复杂制剂的仿制门槛。中国是仿制药大国但高端制剂开发的水平仍然比较薄弱,大多数停留在实验室水平。打破跨国公司对高端制剂的垄断已是迫在眉睫,也是投资创业很好的方向。递送技术的前沿领域智能药物递送系统01基于生理信号或者疾病标志物的智能递送系统是当前热门的研究领域。比如,根据病人的血糖水平控制胰岛素的递送,将血糖稳固在不高不低的正常范围内。早在2015年,浙江大学顾臻教授率先报道了一款新颖的胰岛素响应微针贴片(7),针体是带有硝基咪唑的高分子聚合物,其中包裹有胰岛素和葡萄糖氧化酶。微针接触到血液中的葡萄糖后,氧化酶迅速将其氧化,消耗大量氧气并产生局部缺氧的环境。缺氧条件下,硝基咪唑被还原成氨基咪唑,聚合物的性质发生变化并开始溶解,里面包裹的胰岛素被慢慢释放。微针的长度在 600-800 微米,难以触及皮下的神经细胞,痛感微弱,对皮肤造成的过敏和炎症也很少。针对机械、温度、电流、光磁,酸碱和各种生物标志物的响应微针已经成为智能递送系统的热点(8)。目前,包括顾臻团队研制的智能胰岛素贴片在内的,多个智能微针产品已经进入临床研究。图5:响应微针(responsive microneedle)口服注射装置是解决大分子口服给药的一个替代方案。这种装置大小类似胶囊,患者口服后通过胃肠内壁注射给药,没有强烈的痛感和不适感,最后整个胶囊会通过消化道排出体外。Rani公司用于递送多肽药物PTH的口服注射胶囊已经进入二期临床研究。Biora公司也有类似的技术,用于递送阿达木单抗和GLP-1多肽。最近,MIT教授Robert Langer、Giovanni Traverso和诺和诺德合作开发了一款更加智能的口服注射胶囊(9)。它的外形设计参考了 “豹龟”,拥有一个高且陡峭的外壳,无论以哪种形式进入胃它都能够自己调整方向,把针头对准胃壁。该装置的柱塞和针头由固体糖制成的颗粒控制。柱塞的颗粒首先溶解并将针头下放插进胃壁中,同时柱塞把液体药物挤出。待注射完毕,位于针头之上的颗粒溶解,柱塞会把针头拉回到胶囊中。与之前的类似装置相比,其载药量也实现了数量级提升,每次给药量达到4mg, 因此可以用于疫苗、多肽和蛋白药物等多种生物制剂的口服递送。图6:MIT智能口服注射胶囊的工作原理随着合成生物学、基因工程和光遗传技术的发展,基于小分子和光照等信号响应输出酶、抗体、激素等药物的智能细胞工厂逐渐成为可能。2019 年,华师大生命科学院叶海峰团体报道了以绿茶富含的原儿茶酸为开关,调控工程细胞释放胰岛素治疗糖尿病的研究(10)。2021年,美国纪念斯隆-凯特琳癌症中心的研究人员报道了一种新颖的SEAKER细胞( synthetic enzyme-armed killer cells)技术。SEAKER细胞与肿瘤细胞结合后分泌一种酶,可以水解小分子毒素的前药,实现选择性杀伤肿瘤细胞(11)。自主识别目标和自我复制的 RNA 药物和疫苗也可看作是一种智能的基因递送系统。目前自我扩增RNA (self-amplifying RNA,saRNA)的设计有两种,一种将编码RNA聚合酶和表达目标蛋白的序列装在同一个线性mRNA中,另一方法是将这两种mRNA分开导入细胞中。这种技术的主要优势在于它可以通过自我复制用很低的剂量达到传统mRNA相同的蛋白表达水平。比如,自复制mRNA疫苗在剂量只有传统mRNA几百分之一的情况下能够产生相同的免疫应答。英国初创公司VaxEquity于2021年与阿斯利康达成1.95亿美元的交易,合作开发自复制RNA管线。最近,类似的技术公司Replicate Bioscience宣布完成4000万美元的A轮融资,将致力于开发针对肿瘤、自身免疫和炎症等多种疾病的自复制RNA创新疗法。3D打印和电子药片等智能生产和给药技术也值得关注。3D打印可以智能设计和制造有利于药物吸收的药品形状和三维结构,如2015年上市的抗癫痫药左乙拉西坦口崩片Spritam®。数字健康已是大势所趋,Otsuka在2017年推出一款治疗精神分裂的阿立哌唑电子药片Abilifymycite®。当患者摄入药物后,他们的医生可以轻松追踪信号,因此可以更好地管理和跟踪患者的服药状况。电子药片最终可能发展成为一种诊断、分析和治疗一体化的智能电子给药系统,口服或埋入后能够实时监测疾病指标并根据需求精准释放药物,医生、病人和家属都可以通过手机 APP 等智能终端进行可视化操作。微/纳米机器人02生物相容性材料制造的小型微/纳米装置可以携带药物、诊断试剂甚至活细胞和微生物,并能被化学反应,生物引擎或外部场的作用力所驱动,从而实现药物或者其他载物的运输和释放。制造、驱动和降解是微/纳米机器人(Micro/Nano-Robot)给药系统需要考虑的三个方面(12)。微/纳米机器人的理想制造方法是生物材料的自组装,比如DNA分子通过碱基配对自组装成相关纳米结构。具有管状结构的微/纳米机器人常用化学蚀刻驱动的自卷曲技术制造。对一些结构复杂的微/纳米机器人需要更加先进的制造方法如双光子聚合 3D 打印,但这种方法需要昂贵的仪器和特殊材料。微/纳米机器人的驱动最为关键。化学驱动通常需要催化剂(如活泼金属)和燃料,比如机器人外表的铂金(Pt)催化分解双氧水(H2O2)产生气泡,推动自身的运动。生物驱动主要是利用细菌和精子这些通过鞭毛推动自身运动的微生物作为引擎,这会显著提高微/纳米机器人的生物相容性和安全性。光、超声波或磁场等外部场驱动则不需要燃料,与其他方式相比,它们在控制运动方面更加灵活。比如赤铁矿纳米粒子,依靠表面电荷的吸附与药物结合,能够超声定位并通过磁场操纵其运动。使用可降解材料可以避免移除等后续操作,给微/纳米机器人的应用带来便利并提高其安全性和病人使用的依从性。DNA、多糖、明胶以及水溶性高分子聚合物等材料常用于生产可生物降解的给药微型机器人。微/纳米机器人也可以添加智能化的元件,利用生理信号或生物标记物靶向运输和触发药物释放。加州大学的科学家利用金属镁与酸性溶液反应产生氢气作为动力,推动具有抗肿瘤作用的病毒样颗粒靶向肿瘤组织,在动物模型上显示了显著的效果(13)。国家纳米科学中心主任赵宇亮院士2018年报道了一款DNA纳米给药系统(14)。如图7所示,这项工作利用DNA折纸术(DNA origami)自组装成一个平面结构并通过修饰卷曲成管状装置,其内部包裹凝血酶 (thrombin),外部则连接可以结合Nucleolin蛋白的DNA适配体。Nucleolin在肿瘤血管上皮细胞中高表达,适配体与其结合后管状DNA装置打开,其包裹的凝血酶暴露促进血液凝结,阻断肿瘤组织的血管并抑制肿瘤的生长。这时,DNA适配体既充当靶向肿瘤组织的弹头,也是触发药物释放的开关。图7:抗肿瘤DNA纳米给药系统的工作原理仿生递送系统03为了保证组织、细胞和生命分子之间的交流,生物设计了许多精妙的递送系统。如图8所示,仿生药物递送系统近年来取得了很大的进展,主要载体包括细菌、病毒、细胞、以及外泌体等各种囊泡和生物颗粒(15)。图8:仿生递送系统细菌可以作为活体药物,如最近FDA批准的粪便灌肠药物Rebyota,也可以作为一类仿生递送系统(16)。化学偶联或物理性装载药物理论上可以保留细菌的某些关键属性,例如感知生理信号、自我驱动和组织靶向性等,但面临诸多技术困难,目前仍然探索较少。简化的细菌外膜囊泡(OMVs)进展更为顺利,该技术开发的紫杉醇产品已经进入临床研究(17)。当前,更为热门的技术是基因工程改造的微生物给药系统。CRISPR-Cas等基因编辑工具和合成生物学的发展让工程化细菌的设计和操作变得方便灵活。借助基因工程,可以改变细菌的特征,例如靶向性、粘附能力、蛋白分泌以及特定代谢物的合成,进而成为原位“药物工厂”,在疾病部位生产所需的治疗药物,包括小分子代谢产物、酶、蛋白质和小核酸药物。比如,麻省大学和Ernest公司的研究人员利用三条基因回路的改造分别提升了沙门氏菌对肿瘤细胞的侵袭,裂解和蛋白药物的释放,从而获得了一种专门针对癌细胞输送蛋白药物的、高度改良的细菌递药系统(18)。AAV等病毒是一种常见的核酸物质递送载体,但在递送效率和安全性方面仍有很大的改善空间。哈佛大学George Church等人创立的Dyno通过机器学习和高通量筛选建立了开发全新AAV病毒的研发平台。AI的帮助让他们触及了天然AAV无法到达的序列空间,比如他们从28个氨基酸片段的AAV2序列获得了110,689个变种,数目超过了野生序列的多样性(19)。另一个哈佛教授创立的公司Affinia则是利用新型的AAV9系统开发肿瘤和罕见病药物,目前产品仍处于早期阶段。最近,Flagship孵化的Ring Therapeutics宣布完成1.17亿美元的B轮融资。Ring的核心技术是基于指环病毒(Anellogy)的递送系统,这类病毒在细胞内生命周期较长但不会过度激发免疫反应,使其成为可替代AAV的潜在给药系统。工程化的指环病毒携带单链环状DNA,入核后无法与宿主DNA整合,理论上降低了基因突变的风险。指环病毒的安全性和有效性如果在临床得到验证,将为基因疗法带来新的给药选择。利用细胞载药,尝试较早的是运输氧气的红细胞,作为无核细胞,其载药量大并具有肝脾等网状内皮组织靶向性,体内循环时间长,是理想的药物递送载体之一。Rubius、EryDel、EryTech、西湖生物医药等公司已有产品处于临床阶段。红细胞需要在低渗溶液中膨胀下载药,不可避免有所损伤。Rubius等公司采取干细胞衍生的工程化红细胞,并在红细胞内部表达蛋白药物,避免了低渗负载的方式。西湖生物采取类似的技术路线,并使用外周血来源的造血干细胞,进一步解决了干细胞的来源问题。工程化的血小板也能作为递药载体,比如高效转染遗传物质 (mRNA、siRNA、microRNA、质粒DNA)并将其递送到目标器官,此类技术公司包括Plasfer、PlateletBio和Cellphire Therapeutics等。有意思的是,浙江大学顾臻教授基于血小板靶向出血部位以及在炎症环境下激活的生理特征首创了一种巧妙的肿瘤药物递送系统。最近,该团体尝试将表面携带anti-PD-L1单抗的血小板用于肿瘤手术后的维持治疗,以预防肿瘤的复发和转移,在动物模型上取得很好的效果(20)。顾臻团队最近报道了经液氮处理失活的肿瘤细胞作为药物靶向递送系统的工作,也非常具有新意(21)。顾臻教授作为科学创始人的臻乐医药正致力于推动这些新技术的临床转化,目前已获得超亿元的融资支持。细胞外囊泡(EVs)是细胞通讯交流的基本工具,有着极其重要的生理功能。细胞分泌的囊泡结构上类似于人工合成的脂质体但保留了许多来自母细胞的调控分子和特征,相较于传统载体有归巢功能,以及低毒、低免疫原性和高效等诸多优势。外泌体是当前研究最多的天然囊泡,近年来被广泛应用于诊断和药物递送。外泌体的生产、提纯和质量控制是阻碍其向临床转化的最大障碍。纳斯达克上市公司Codiak发现PTGFRN和BASP1是外泌体组装的关键分子,通过与这两个骨架蛋白的融合可以将需要递送的药物搭载在外泌体的外侧或内侧。这种工程化的外泌体技术采取内源表达的药物装载方式,可以使用色谱和过滤代替传统的超速离心,大幅提高了GMP生产的效率和纯度。Codiak构建了多个针对肿瘤和神经疾病的药物管线,临床产品包括肿瘤免疫药物exo-STING 和exo-IL-12等。另有多家外泌体公司获得了大额投资以及与跨国公司合作的机会,包括Evox,ArunA Bio以及国内的唯思尔康和恩泽康泰等。除了囊泡,各种生物颗粒也成为药物递送探索的目标。2023年2月份,张锋创立的拥有全新病毒样颗粒(VLP)递送技术的基因编辑公司Aera Therapeutics获得了近两亿美元的B轮融资。该公司的核心技术就是张锋团队2021年发表在《科学》杂志上的一项工作(2)。他们发现逆转录病毒样蛋白PEG10能够与mRNA结合并在其周围形成球型颗粒,通过优化和改造成功地开发了一种包装和递送mRNA的通用系统(SEND)。该系统来自人源病毒,与现有的病毒载体和脂质纳米颗粒相比,所引起的免疫反应更小,并且能够将基因编辑工具高效地递送到小鼠和人类细胞中并完成目标基因的编辑。无独有偶,刘如谦实验室研究了影响VLP递送效率的几个特性,然后通过工程化改造优化了其生产、携带和释放等步骤(3)。新的VLP系统可以高效地将基因编辑工具递送到动物体内的细胞中进行相关疾病的治疗。2023年3月份,张锋团队在《自然》杂志上报道了另一项革命性的、类似于细菌可收缩注射系统PVC-eCIS的蛋白递送技术,引起媒体和投资者的极大关注(4)。许多病原菌拥有一类能够将毒素等蛋白大分子递送到宿主细胞内的可收缩注射系统,包括六型分泌系统T6SS和发光杆菌毒力基因簇PVC等类型。其中,分泌到细菌胞外的PVC颗粒便于制备且不具有传染性,尤其受到关注。如图8所示,PVC的工作原理非常类似于注射器,尾部丝状蛋白识别并固定于细胞膜上,而外鞘的弹性蛋白收缩推动针头状的装置穿透细胞膜并将所载蛋白注入胞内。在张锋工作发表之前, 中国医学科学院江峰/金奇团队在2022年4月发表了一项开拓性工作,他们鉴定了一类N端信号肽,携带这个信号肽的蛋白分子能够导入PVC颗粒并被转运至真核细胞内,解决了PVC作为递送系统的药物装载问题(22)。在此基础上,张锋团队则发现尾丝蛋白Pvc13与细胞膜受体的相互作用是PVC颗粒识别目标细胞的关键。利用最新的AI技术对Pvc13进行蛋白工程改造,可以让PVC识别表达EGFR等受体的人源细胞,这项工作进一步解决了PVC的靶向问题,并让人体应用变得可能。至此,改造后的PVC可实现体内高效递送Cas9和毒素等功能蛋白,为基因编辑和疾病靶向治疗提供了全新的解决方案。当然,这类细菌来源的生物颗粒是否存在免疫原性,其递送效率和安全性是否具有优势仍需大量探索工作。图9:最近报道的新型仿生递送系统仿生递送系统虽然极具前景,但技术复杂,大多数仍处于探索阶段。最近,Codiak以及红细胞递送公司Rubius的临床产品数据不佳,导致这两个曾经的明星公司陷入困境。这也让许多人对这类仿生技术产生了质疑。在下结论之前,有几点值得思考。首先,不同于传统的改良新药,Codiak产品Sting,IL-12和Rubius的IL-15都是全新的肿瘤免疫药物,靶点本身风险极大,产品失败不能完全归结于递送系统。其次,两个公司都是采取内源表达的载药方式,药物和递送系统融为一体,成为一种全新的药物形式。类似于ADC,这类药物和递送系统的组合通常会涌现一些新性质。如果缺乏对靶点的深刻理解和充分的评价,这种独特的药物形式是否有利于治疗往往不可预测。因此,拥有这类技术的公司不能为了递送而递送,而是要耐心找到可评价且真正解决问题的切入点。最后,任何新技术都不是一蹴而就的,只有依靠时间和实践的检验才能准确认识,仿生递送仍然非常值得期待。核酸的肝外递送04前面提到,核酸药物和疫苗的快速发展得益于脂质纳米颗粒(LNP)和GalNac偶联技术的日益成熟。然而,现有技术特别是GalNac只能高效靶向肝脏,让核酸药物的潜力不能充分释放。因此,有人提出核酸药物的下个突破就是解决肝外递送的问题。LNP虽然常在肝脏中累积,但研究表明修改LNP的内外部电荷或添加额外的分子,能够改变LNP原有的肝靶向。2020年,美国西南医学中心 Daniel J. Siegwart团队开发了一种被称为选择性器官靶向(SORT)的技术,通过添加新的脂质分子调节LNP的摩尔组成和内部电荷,实现了肝、肺和脾等不同组织的RNA特异性递送(23)。RNA内吞进入细胞后一般卡在内吞体(endosome)当中,只有少部分能够逃逸进入胞内发挥作用。Siegwart团队2021年又开发了一种由新型磷脂(iPhos)组成的、具有内吞体逃逸性能的新型LNP递送系统(iPLNPs)。该系统显示了极高的体内mRNA递送和CRISPR/Cas基因编辑效率,而且通过iPhos化学结构和比例调控可实现器官选择性递送(24)。基于这些技术的初创公司ReCode最近获得Pfizer领投的大额投资,致力于开发靶向气管上皮细胞的基因编辑药物,治疗遗传性呼吸系统疾病PCD和CF。图10:ReCode 的器官靶向SORT-LNP(25)另外,美国塔夫茨大学许巧兵团队发现尾部含有酰胺键的新型LNP 能够选择性地将mRNA传递到小鼠肺中,而且只要简单调整LNP的头部结构就可以靶向不同的肺细胞类型(26)。最近,宾夕法尼亚大学的研究人员设计合成了一系列新颖的可离子化脂质,最终发现了一种能够高效递送mRNA到胎盘而不进入胎儿体内的LNP颗粒,有望用来治疗孕期并发症先兆子痫(27)。2021年,初创公司GenEdit宣布完成2600万美元的A轮融资,专注开发非病毒、非脂质聚合物纳米粒子递送载体,并针对神经系统疾病开发临床候选药物。该公司的NanoGalaxy平台可以依据不同货物分子修饰新型聚合物的结构从而实现DNA、RNA或CRISPR蛋白复合体的组织选择性递送。Altamira开发的OligoPhore™ / SemaPhore™ 是一种由21个氨基酸多肽和小核酸或mRNA压缩形成的聚合物。这种颗粒的形状和携带的电荷让其避免了肝脏的快速清除,因此可以到达其他组织和器官。Entos的Fusogenix技术是一种由中性脂质和专有的FAST(Fusion-Associated Small Transmembrane)蛋白组合形成的递送系统,可以高效地将核酸物质和其他药物递送到目标细胞和器官。Genprex的平台技术ONCOPREX®是一种带有正电的纳米囊泡,静脉注射后带负电的肿瘤细胞选择性地摄取这些纳米颗粒并表达携带的抑癌基因比如TUSC2。2020年美国FDA批准了Genprex这款基因药物的临床申请,与第三代TKI奥希替尼联用治疗非小细胞肺癌。有别于GalNac的新型偶联也是解决肝外的方向之一。Alnylam的研究人员最近发现O-十六烷基(C16)修饰的siRNA能够进入中枢神经系统(CNS)、眼睛或肺部并产生长达3个月的基因敲除作用(28)。DTx公司脂肪酸修饰的FALCONTM平台非常类似,可以靶向不同的肝外组织。他们开发的第一款Falcon-siRNA DTx-1252靶向神经外围组织Schwann细胞中过表达的PMP22基因,用于治疗一种不算十分罕见的神经肌肉疾病CMT1A。2023年7月17日,诺化宣布首付和里程碑各5亿美元收购临床前产品DTx-1252 和该公司其他早期资产的某些权益。Avidity开发了一款抗体偶联药物AOC1001,利用TfR1单抗将降解DMPK的siRNA带至肌肉,用于治疗1型肌营养不良症(DM1)。2022年12月份Avidity公布了I期阳性数据,股价大涨。另外,Entrada公司的小环肽递送系统(EEV™)也宣称可以有效加强药物的内吞体逃逸。该公司2021年成功登陆纳斯达克,募集资金主要用于其EEV偶联核酸药物ENTR-601-44和ENTR-501的临床研究。局部给药也可一定程度上实现器官的靶向性。2022年12月Vertex和Moderna合作研发的VX-522治疗囊性纤维化(CF)的IND申请获得FDA批准。VX-522将全长CFTR mRNA封装在LNP并通过吸入的方式递送至肺部。今年1月,Arcturus利用其专有LUNAR递送技术开发的、用于治疗CF的吸入式mRNA疗法ARCT-032获得IND批准并将在新西兰进行首次人体实验。enGene的双衍生壳聚糖(DDX)可以高效地舌下递送基因(plamid DNA)。其他还有Ichor开发的TriGrid®,则是在目标器官使用自动化的电穿孔技术,大大提升局部核酸物质的吸收。还有一种间接的策略是在给药前注射一种暂时占据肝脏的脂质体(Nanoprimer),从而提高其他组织对含药LNP的吸收(29)。研究表明,用Nanoprimer对小鼠进行预处理可提高包裹人促红细胞生成素(hEPO) mRNA或VII因子(FVII) siRNA 的LNPs的生物利用度,分别导致hEPO的产生增加32%或FVII的沉默增加49%。新兴的偶联递送技术05ADC的成功让偶联药物重新成为热点,各种新技术呈现出“万物皆可偶联”的态势。偶联技术的创新可以简地分为三类,即载荷(payload)、连接子(linker)和靶向端(targeting)。载荷除了常规毒素外,还包括各种新毒素、抗体免疫刺激偶联药物(ISAC)、抗体蛋白融合药物(APC)、抗体细胞偶联药物(ACC)、抗体寡核苷酸偶联物(AOC)、抗体生物聚合物偶联物(ABC)、抗体降解剂偶联(ADeC)以及当前非常热门的放射性核素偶联药物(RDC)等。需要注意的是,激素,小分子免疫激动剂以及IL-2等免疫偶联药物临床开发并不顺利。原因可能多种多样,生物学上值得研究的是,这类免疫药物通过偶联递送局限于微环境是不是正确的方向。RDC的出发点与ADC类似,采用靶向偶联技术减少对正常组织的杀伤。其载荷是螯合物(Chelator)和金属放射性核素配位形成的复合物,通过连接臂与靶向分子相连,构成完整的RDC药物。核素作为载荷有诸多优点,如可以显像,集生物标志物、诊断和治疗于一体。然而,射线的杀伤作用不能像化学毒素一样,通过形成偶联药物暂时被掩盖。这一特点导致RDC的设计与ADC有重大差异。最新的ADC多采用肿瘤组织特异性切割的连接子,可以进一步增加药物的靶向性。RDC则不同,一旦进入体内其携带的核素始终保持发射杀伤性射线直至排泄体外。不难理解,目前上市和在研的RDC药物多采取半衰期较短的小分子和短肽作为靶向分子,如诺华治疗前列腺癌的Pluvicto(77Lu-PSMA-617)使用的就是靶向PSMA的小分子(图11)。单抗的靶向性更好,但长达几周的半衰期容易导致严重的副作用。RDC的Linker与ADC也有所不同,不需裂解,可以将其作为RDC的一部分来考虑。图11:Pluvicto的化学结构和作用原理硼中子俘获治疗技术(BNCT)近年来逐渐受到关注。这种疗法的原理是首先将携带稳定同位素10B的化合物特异性地集聚在肿瘤组织内,随后用中子束对肿瘤部位进行照射发生硼中子俘获作用释放α粒子和7Li粒子杀伤肿瘤细胞。这种分两步走的技术理论上可以解决核素毒性难以暂时掩盖的难题。然而,BNCT不但需要配套昂贵的中子发生器,还需高浓度的硼化合物才能产生足够剂量的射线,显然通过抗体偶联等方法难以实现硼的富集。目前在日本获批的、治疗头颈癌的BNCT药物BPA是具有一定肿瘤靶向性的小分子硼化合物。通过化学改造开发更加稳定、高效和水溶性改善的裂解或不可裂解的Linker也是偶联药物创新的重要方向,文章和专利多有报道,这里不再赘述。新兴的偶联技术在靶向端的创新包括新型ADC(双抗,Probody等)、病毒样颗粒偶联物(VDC),各种高分子载体、抗体片段偶联药物(FDC),适配体偶联药物(ApDC)和基于点击化学的偶联技术等。双抗和条件激活抗体(probody)是ADC药物常见的差异化设计。最近,基于DS-8201的成功,多款双抗与喜树碱类毒素偶联的ADC 进入临床研究并获得了初步阳性数据,包括Her2/Her3,Her3/EGFR , c-Met/EGFR等。CytomX和AbbVie共同开发的Probody偶联药物CX-2029也进入了临床研究,但早期数据相当负面,在16例NSCLC患者中客观缓解率(ORR)仅为18.8%。Xilio公司所谓的肿瘤微环境选择性激活的单抗和炎症因子药物2021年也已进入临床研究,暂未见临床数据报道。比利时Complix公司的Alphabody穿膜抗体技术可以让单抗等大分子靶向细胞内的靶点。Alphabody是一种由三个α螺旋特殊结构组成的大小仅10kDa的非天然蛋白,可以携带药物分子有效穿过细胞膜。2017年拉斯克奖获得者John T. Schiller等人发现HPV病毒颗粒能特异地与肿瘤细胞表面的硫酸乙酰肝素蛋白聚糖(HSPG)结合,因此可以应用于类似于ADC的偶联递送。Schiller创立的Aura公司2021年获得8000万美元的投资并成功登陆纳斯达克,其第一款药物AU-011使用了一种可被红外光激活的强效细胞毒药物作为载荷。被激活后,AU-011可产生高水平的单线态氧,选择性破坏肿瘤细胞并激活免疫系统产生抗肿瘤反应。肿瘤的血管通透性更高,分子量较大的物质被认为具有EPR效应,即更容易进入肿瘤组织并长期滞留。因此,高分子材料常用来作为肿瘤药物的靶向载体,但临床一直未能重现动物试验观察到的良好靶向效果。高分子载体与抗体等靶向性分子进一步偶联是最新的改进方向。2022年12月份,美国加州的Dantari公司宣布完成了4700万美元的A轮融资。他们的核心技术是高载荷抗体偶联药物(T-HDC),通过携带多个药物分子的高分子载体与抗体偶联,最终DAR值可高达60,远高于常规偶联药物2-8的DAR值范围,因此适用于毒性更低的载荷。Dantari通过表面化学修饰减少这一类超高分子量药物的免疫原性并降低了其骨髓暴露量。T-HDC技术覆盖的有效载荷包括拓扑异构酶抑制剂、紫杉烷类、蒽环类药物,靶点涉及HER2、PSMA、Trop-2、HER3、DLL3等。总部位于新泽西的Elucida Oncology则是利用康奈尔大学教授Uli B. Wiesner开拓的,一种直径在5-6纳米的超小型硅胶颗粒(C'Dot)作为药物偶联的载体。这种小型的C'Dot体积只有ADC的三分之一,能够更好地穿透肿瘤组织并能通过肾排泄清除,携带的药物分子可以高达80个而不影响其理化性质。该公司C'Dot偶联技术开发的第一个药物ELU001,在硅胶核的表面分别携带靶向叶酸受体α的小分子和喜树碱类似物exatecan,已经进入临床研究,用于治疗包括卵巢癌在内的恶性肿瘤。点击化学(Click chemistry)和生物正交化学(bioorthogonal chemistry)获得了2022年度诺贝尔化学奖。这类化学反应的特点是反应简单高效且不影响生物体内的正常生物化学反应,因此广泛应用于生物探针和诊断等领域。有意思的是,这类反应最近被发现可用于新型偶联递送技术的开发。西湖大学工学院院长程建军教授是较早的开拓者之一,他任职美国伊利诺斯香槟分校时报道了一项极具创造性的药物递送技术ATTACK(Active Tissue Targeting via Anchored Click Chemistry)。如图12所示,该技术用一种含有叠氮的代糖(Ac4ManNAz ,N-叠氮乙酰甘露糖胺)修饰和标识细胞表面,因为肿瘤细胞嗜糖的Warburg效应其表面叠氮的含量远远高于正常细胞。含有炔类分子DBCO的偶联药物可以识别叠氮并通过点击化学将药物不可逆地靶向肿瘤细胞,随后以类似于ADC酶切的方式在肿瘤组织特异性释放毒素或其他药物(30)。ADC药物虽然取得了很大的成功,但大部分肿瘤并没有类似于Her2这样的特异性高表达靶标。ATTACK技术实际上在肿瘤组织中人工创造了一个有别于正常组织的靶标,因此,非常巧妙地为无法靶向肿瘤的治疗提供了一种解决方案。程建军教授创立的苏州瑞奥生物目前已经获得数千万融资,利用 ATTACK 技术开发独特的抗肿瘤药物,公司预计在 2024年递交首个IND。图12:ATTACK技术的工作原理成立于2021年的生物医药公司Shasqi开发了一种点击化学触发的药物释放技术。如图13所示,该技术将阿霉素与反式环辛烯(transcyclooctene,TCO)连接为一种前药,给药之前在肿瘤部位注射含有四嗪(tetrazine)的高分子,这样TCO前药到达肿瘤微环境后快速与四嗪发生环加成反应并释放阿霉素,从而实现了可控的、肿瘤特异性的药物释放(31)。2023年7月份,Nature 生物技术子刊特别介绍了Shasqi与强生公司的合作极其新型阿霉素偶联药物的临床进展(32)。图13:点击化学触发的药物释放透脑的递送技术06最近,渤健(Biogen)和卫材(Eisai)联合开发的阿尔茨海默病药物lecanemab获得FDA正式批准,预示着中枢神经系统药物可能成为下一个热点。开发中枢药物的一个挑战是透过血脑屏障,特别是分子量较大的药物,常常需要靶向中枢的递送系统。最近,武田与PeptiDream达成35亿美元合作,开发治疗神经退行性疾病的多肽偶联药物,其递送策略是利用BBB高表达的人转铁蛋白受体1(TfR1)的转运作用。Flagship参与投资的Denali也拥有一项类似的穿透血脑屏障的靶向转运平台(ATV)。该公司临床II期产品DNL310是一款IDS酶和靶向TfR1抗体的融合蛋白药物,借着TfR1转胞作用(transcytosis)将IDS递送到大脑,用于治疗因为IDS基因突变引起的一种罕见的神经退行性溶酶体贮积症。图14:Denali的CNS靶向转运平台ATVLNP、外泌体和AAV均有脑部递送包括小核酸在内的各种药物的成功案例,如uniQure的AMT-130就是由AAV5将microRNA递送到大脑中抑制突变亨廷顿蛋白的产生,以治疗亨廷顿舞蹈症(HD)。Bioasis Technologies开发了一种多肽偶联递送技术,利用他们专有的肽段促进siRNA的转胞内吞作用,从而穿越血脑屏障。此外, 纪念斯隆·凯特琳癌症中心的科学家最近报道了一种能够突破血脑屏障的靶向P-选择素的岩藻多糖-纳米颗粒,利用P-选择素的转运将小分子抗肿瘤药物vismodegib递送到脑肿瘤组织中,显著提高了药物的治疗效果(33)。体内细胞疗法的递送技术07以CAR-T为代表的细胞疗法取得了巨大成功,然而个性化导致的高成本限制了其应用。这恰恰也是下一代细胞疗法的突破方向。通用型CAR-T、iPSC技术,以及新的生产技术如“cell squeeze”(SQZ)等都在某些程度上可以降低成本。然而,这些技术路线的进展不尽如人意。最近,基于递送技术的体内细胞疗法逐渐进入人们的视野。传统的CAR-T实际上是一种体外改造的基因疗法。首先抽取病人的原始T细胞,经过一系列的纯化、激活、转染、扩增、冻存、运输等步骤,最后将基因改造的CAR-T细胞输入患者体内。体内CAR-T给患者输注DNA或mRNA ,直接在体内构建或重编程T细胞,使得人体内的T细胞自己转变成CAR-T。由于避免了体外生产的过程,体内CAR-T的成本大幅度降低。而且,不存在细胞来源问题,因此没有异体带来的免疫排斥反应,有望成为一种真正的通用型细胞疗法。从技术的角度看,体内CAR-T就是基因疗法,无非就是将体外转染T细胞这一步放在了体内。然而,体内转染对递送技术提出了更高的要求,需要选择性地将CAR基因或mRNA递送到T细胞当中表达。与传统基因疗法类似,体内CAR-T的递送技术也可以分为病毒和非病毒两种类型。另外,因为不能像体外CART一样使用化疗清淋预处理,为了保证CAR-T体内扩增,递送中还要包含促进T细胞增殖这一环节。病毒递送的代表性产品是Umoja Biopharma “VivoVec“平台开发的临床产品CD19 CAR-T UB-VV100。该技术使用慢病毒(LV)作为载体,携带可以表达CD19和共刺激信号分子4-1BB的基因片段,其表面含有anti-CD3的抗体片段,因此可以靶向和激活T细胞。有意思的是,其LV载体同时携带雷帕霉素激活的细胞因子受体(RACR)。同时服用小分子药物雷帕霉素时,患者正常免疫细胞被抑制而含有RACR受体的CAR-T细胞受到刺激释放IL-15/IL-2反而快速增殖(34)。EXUMA和Mustang 等公司也有类似的技术,他们使用LV携带表达CAR或TCR的基因片段并辅以促进T细胞扩增的方法。EXUMA的方法是同时携带刺激T细胞的信号分子,而Mustang则是使用多肽药物驱动患者T细胞的增殖。2022年8月份成立的体内CAR-T公司Vector的递送技术是苏黎世大学教授Andreas Plückthun实验室开发的基于腺病毒血清型5(Ad5)的SHREAD(SHielded, REtargeted ADenovirus)基因递送系统(35)。SHREAD通过DARPins分子修饰将Ad5重新靶向那些表达选定标记物(如Her2,CD3等)的特定细胞,并在靶细胞高效表达所携带的基因,而且SHREAD使用了基于抗体三聚体单链可变片段(scFv)的防护罩,可大大降低递送系统的肝脏嗜性和免疫原性(36)。早在2017年,西雅图Fred Hutchinson癌症研究中心的科研人员就报道了利用修饰的LNP递送系统对人体中的T细胞进行重编程,转变为CAR-T的方法。该技术也是利用anti-CD3e f(ab′)2片段修饰赋予LNP免疫细胞靶向性,同时通过细胞核定位多肽MTAS和NLS确保所包裹的CAR基因能够进入细胞核(37)。英国初创公司Ixata采取了一种类似的,CD3靶向适配体修饰的LNP包裹慢病毒的递送路线,LNP封装的慢病毒载体直接注射到患者的血液中并改造患者体内的免疫细胞。2022年,美国宾大科学家在《科学》杂志上发表了一项开创性工作,他们向小鼠体内注射mRNA,对患心衰的个体进行体内T细胞的改造,成功修复了小鼠心脏的功能(38)。Juno的创始人,CAR-T疗法的先驱Carl June创立的Capstan公司2021年获得了6300万美元的投资,他们的核心技术也是通过LNP-mRNA 在体内直接改造T细胞。2022年,巨噬细胞新锐Carisma宣布与mRNA巨头Moderna合作,开发基于mRNA的体内CAR-M技术。国内的远泰生物在2022 AACR上发布了其mRNA-LNP在CAR-T/NK领域的最新研究进展。Juno前高管团队打造的Sana公司A轮获得了7亿美元融资,创下细胞疗法领域单笔最高融资记录,他们通过收购Cobalt公司获得了一种全新的体内细胞疗法的递送平台——Fusogen技术。Fusogen是一种来自病毒由受体识别G蛋白和膜融合F蛋白组成的,介导细胞间和细胞内融合的天然蛋白复合物。Fusogen能够与目标基因形成囊泡Fusosome,然后融合进目标细胞并进行基因组整合,从而实现体内细胞的重编程。通过对G蛋白进行改造,如添加scFvs、VHHs或DARPins,可以获得识别不同细胞类型(包括T细胞、肝细胞或造血干细胞)高度选择性的Fusogen递送系统。目前,Sana公司基于Fusogen技术已经开发了多个临床前管线,适应症涵盖了肿瘤和罕见病等。其中Fusogen CD19 CAR-T项目在小鼠试验中取得较为积极的结果,通过IV给药Fusosome体内构建CD19 CAR-T可以清除人源化小鼠体内的B细胞肿瘤。图15:Sana的Fusogen递送平台活体药物的递送技术08细菌、细胞和病毒等活体药物本身可以口服,埋入或注射给药。然而,由于他们具有生命特征,递送系统也常常需要特殊设计,比如避免体内环境对药物的破坏作用。水凝胶、微生物薄膜(MBT),聚合物涂层等是常见的支持细菌存活的递送系统。上海交通大学分子医学研究院刘尽尧教授近年来报道了一系列细菌药物的隐身策略,包括在细菌的表面添加稳定的保护层如红细胞膜、脂质体等,或者诱导细菌分泌细胞外基质形成天然的生物膜,这些封装技术可以使细菌药物对巨噬细胞吞噬、胃酸和胆汁酸等破坏性因素产生良好的抵抗能力(39)。近日,该团队报道了一种更加智能的细菌药物递送方法。类似于肠溶胶囊,他们用肠溶性聚合物形成的纳米涂层对细菌进行包裹,细菌在胃部暂时失活而进入肠道时其纳米“外衣”溶解释放活性菌并发挥治疗功效(40)。细胞药物有时也需要支持细胞生长并模拟细胞外基质(ECM)的给药系统,比如治疗软骨损伤的、基质诱导的自体软骨细胞移植技术(MACI)。化学改造的海藻作为植入物可以封装治疗1型糖尿病的诱导胰岛素分泌的β细胞,最大限度地改善细胞生存的微环境以延长β细胞的活力和维持血糖水平。类似的系统可保护并递送经基因工程改造以分泌因子VIII203的上皮细胞,目前,处于血友病治疗的I期临床试验(SIG-001)。2022年8月,北卡罗来那大学的研究人员报道一种名为MASTER(Multifunctional Alginate Scaffold for T Cell Engineering and Release) 的“体内“CAR-T技术(41)。与前面提到的直接改造体内T细胞的方法有所不同,该技术首先也需要从患者身上分离出T细胞,然后将未激活的T细胞、装载CAR的病毒载体、刺激T细胞的抗体以及IL-2进行混合并植入一种多功能藻酸盐支架。MASTER支架植入体内后,病毒首先将T细胞转化为CAR-T细胞,抗体和IL-2开始渗出并促进CAR-T细胞的快速增殖。这相当于将传统CAR-T体外制造工厂搬到了体内,将其生产时间缩短到1天。有意思的是,科学家们在淋巴瘤小鼠模型中发现MASTER技术生产的CAR-T细胞比传统方法表现出更好的疗效。图16:MASTER技术和传统CAR-T的比较溶瘤病毒(Oncolytic Virus,OV)是一类重要的肿瘤免疫治疗药物,但一般需要瘤里注射给药,限制了其临床应用。因此,发展新的能够实现系统给药的递送技术成为下一代溶瘤病毒开发的方向,技术路线主要包括使用细胞和细菌作为载体或者通过外泌体、脂质体,高分子和多肽包裹等。表4:溶瘤病毒新型递送技术(来自网络媒体)结 语随着人类社会的老龄化,越来越多的人需要对各种疾病进行治疗和长期护理。满足当前和未来的临床需求持续要求生物医药新技术的突破,而药物递送是其中最底层的“卡脖子“技术之一。在这次抗新冠的应对中,前药、联用P450抑制剂以及LNP递送mRNA等递送技术发挥了至关重要的作用,为我们快速提供了多种治疗药物和预防疫苗。经过70多年的发展,递送技术已经同时拥有老药改良和新技术赋能的双重功能。无论是改良型高端制剂,还是智能、仿生和偶联等新技术都需整合多元化的学科知识和技能,必将成为未来生物医药创新的必争之地。参考文献1. Nat Rev Drug Discov 2023
May;22(5):387-409(Robert Langer等人最新的综述)2. Science 2021 Aug
20;373(6557):882-889(张峰开发的SEND技术)3. Cell 2022 Jan 20;185(2):250(刘如谦开发的eVLP技术)4. Nature 2023 ,616(7956):357(张峰开发的PVC-eCIS技术)5. Nat Biomed Eng 2021Sep;5(9):951-967(Samir Mitragotri等人最新的综述)6. 谢雨礼博士 | 生物科技的下个10年:RNA药物“王者归来”(药时代)7. PNAS,2015,112(8260),(顾臻教授开发的胰岛素智能贴片)8. Pharmaceutics 2023 May4;15(5):1407(最新的微针综述)9. Nature Biotechnology 2022,40(1):103(MIT的智能口服注射胶囊)10. Sci Transl Med 2019 Oct23;11(515):eaav8826(叶海峰教授报道的可控胰岛素释放)11. Nat ChemBiol 2022 Feb;18(2):216-225(SEAKER细胞技术)12. Journal of IntegationTechnology 2012,10(3),78-92(微/纳米机器人综述)13. Small, 2020, 16(20): 1907150(加州大学肿瘤靶向的纳米机器人)14. Nature biotechnology2018,36(3):258(赵宇亮院士报道的DNA纳米机器人)15. Accounts of Chemical Research2019, 52, 1255(仿生递送系统综述)16. Adv. Mater.2021, 33, 2102580(细菌药物递送的综述)17. PLoS One 10 (2015), e0144559(细菌囊泡包裹的紫杉醇递送)18. Nat Commun 2021 Oct21;12(1):6116(基因改造的沙门氏菌药物递送系统)19. NatBiotechnol 2021 Jun;39(6):691-696(George Church人工智能设计AAV)20. Nature Biomedical Engineering 2017,1, 0011(顾臻教授报道的血小板-PD-L1递送)21. Adv Mater 2023Jul;35(28):e2212210(顾臻教授报道的失活肿瘤细胞递送系统)22. Sci Adv 2022 Apr 29;8(17):2343(江峰/金奇团队报道的PVC装载方法)23. NatNanotechnol 2020 Apr;15(4):313-320(美国西南医学中心的SORT-LNP)24. Nat Mater 2021May;20(5):701-710(改进的SORT-LNP)25. Nature Protoc 2023, 18(1):265(SORT-LNP的制造步骤)26. Proc Natl Acad Sci U S A 2022Feb 22;119(8):e2116271119(许巧兵团队的LNP)27. J Am Chem Soc 2023 Mar1;145(8):4691-4706(宾夕法尼亚大学的LNP)28. Nat Biotechnol 2022Oct;40(10):1500-1508(Alnylam的新型siRNA偶联递送)29. Nano Lett 2020 Jun10;20(6):4264-4269(间接肝外递送技术Nanoprimer)30. Nat Chem Biol 2017 Apr;13(4):415-424(程建军教授开发的ATTACK技术)31. bioRxiv 2023.03.28.534654(Shasqi化学触发的药物释放技术)32. Nat Biotechnol 2023.7.5.DOI:10.1038/s41587-023-01860-2(Shasqi与强生合作的报道)33. Nat Mater 2023Mar;22(3):391-399(P-选择素介导的透脑性岩藻多糖-纳米颗粒)34. Mol Ther Methods Clin Dev 2023 Feb 26;29:120-132(Umoja体内CART技术)35. Mol Ther Methods Clin Dev 2023 Feb 26;29:120-132(Vector体内CART技术)36. PNAS 2021 May25;118(21):e2017925118(苏黎世大学的SHREAD基因递送技术)37. Nat Nanotechnol 2017Aug;12(8):813-820(LNP递送的体内CART技术)38. Science2022 Jan 7;375(6576):91-96(治疗心衰的体内CART技术)39. Nat. Commun., 2019, 10, 3452;Nat. Commun., 2019, 10, 5783;Sci. Adv.,2020, DOI: 10.1126/sciadv.abb1952;(刘尽尧教授细菌药物隐身策略的系列报道)40. Adv. Mater., 2020, DOI:10.1002/adma.202002406(刘尽尧教授智能细菌药物递送系统)41. Nat Biotechnol 2022Aug;40(8):1250-1258(北卡罗来那大学的体内CART工厂MASTER)精彩推荐谢雨礼博士 | 药物创新:Close Is Everything谢雨礼博士 | 药物创新:more is different谢雨礼博士 | 未来十年新药研发,我们要做什么?点击这里,欣赏谢雨礼博士精彩美文!
信使RNA疫苗核酸药物基因疗法
100 项与 CMT1A(溪砾科技) 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 腓骨肌萎缩症 | 临床前 | 中国 | 2024-06-19 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
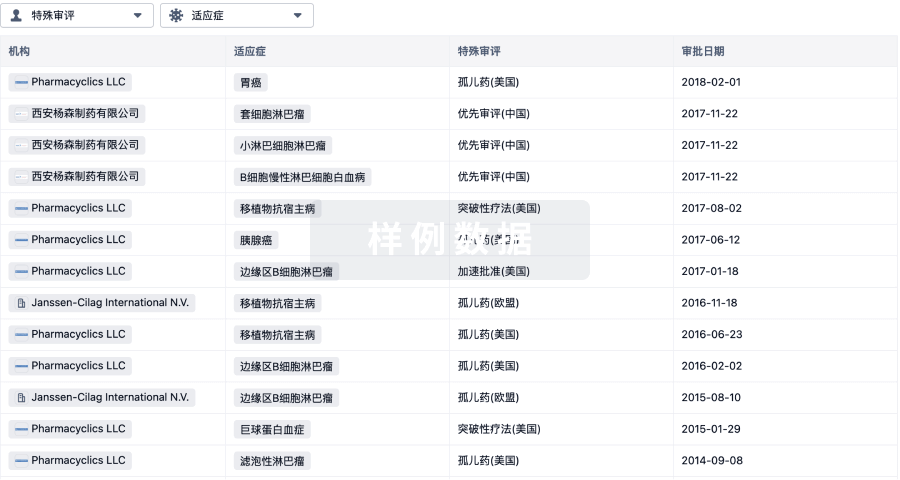
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用