更新于:2024-11-21
P-Vax
更新于:2024-11-21
概要
基本信息

关联
100 项与 P-Vax 相关的临床结果
登录后查看更多信息
100 项与 P-Vax 相关的转化医学
登录后查看更多信息
100 项与 P-Vax 相关的专利(医药)
登录后查看更多信息
36
项与 P-Vax 相关的文献(医药)2024-11-12·Infection and Immunity
Molecular mechanisms of
Coxiella burnetii
formalin-fixed cellular vaccine reactogenicity
Article
作者: Butler, S. M. ; Fratzke, A. P. ; Samuel, J. E. ; Schaik, E. J. van ; Szule, J. A.
2024-03-01·Advanced Healthcare Materials
Synthetic Particulate Subunit Vaccines for the Prevention of Q Fever
Article
作者: Marsh, Ian ; Plain, Karren ; Stenos, John ; Rehm, Bernd H. A. ; Graves, Stephen R. ; Islam, Aminul ; Chen, Shuxiong ; Sam, Gayathri ; Westman, Mark E.
2023-12-28·The Journal of Physical Chemistry A
Computational Risk Assessment of Persistence, Bioaccumulation, and Toxicity of Novel Flame-Retardant Chemicals
Article
作者: Pandit, Shraddha ; Singh, Prakrity ; Sinha, Meetali ; Yadav, Dhvani ; Parthasarathi, Ramakrishnan
100 项与 P-Vax 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 假单胞菌感染 | 临床前 | 西班牙 | 2023-04-21 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
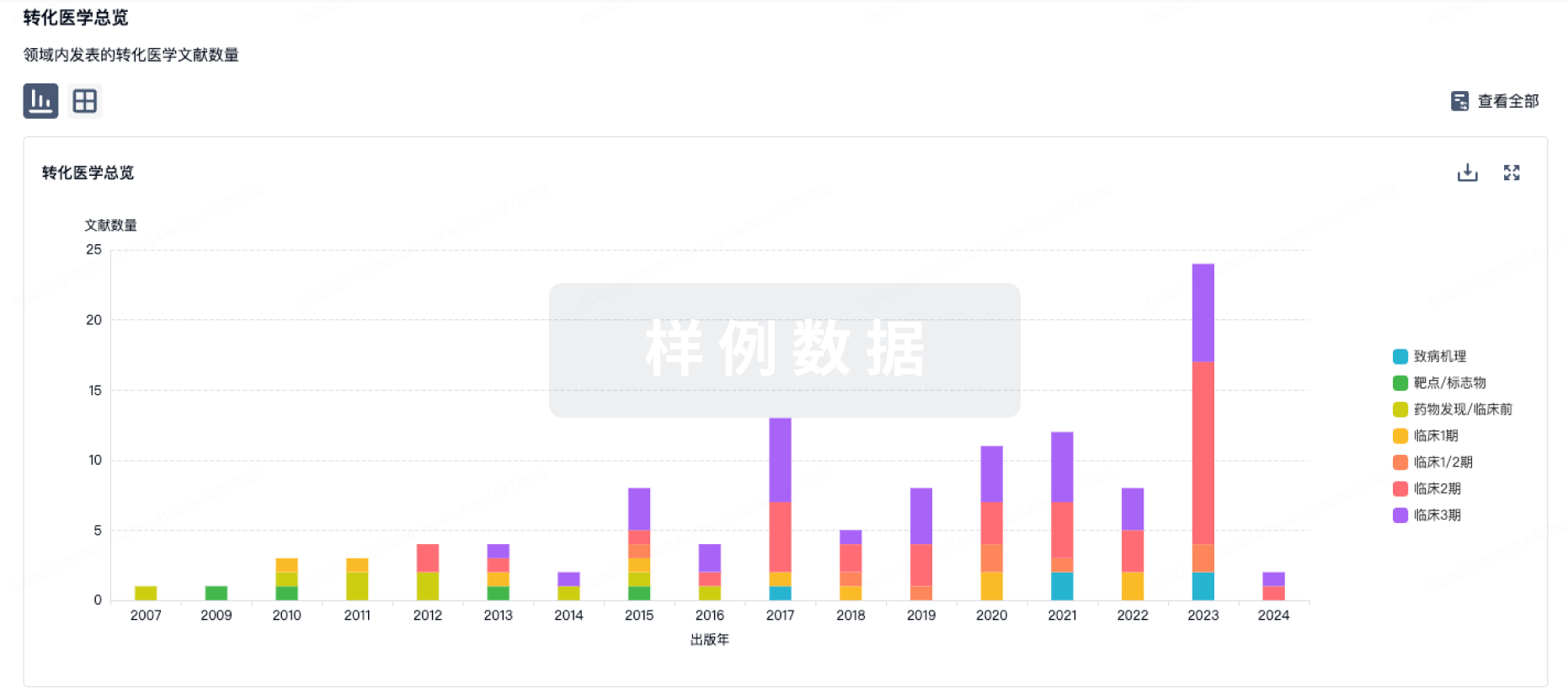
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
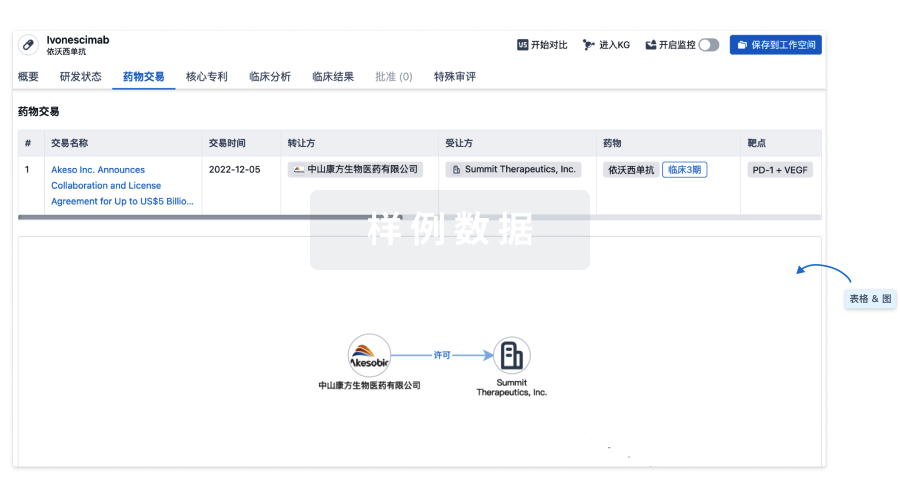
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
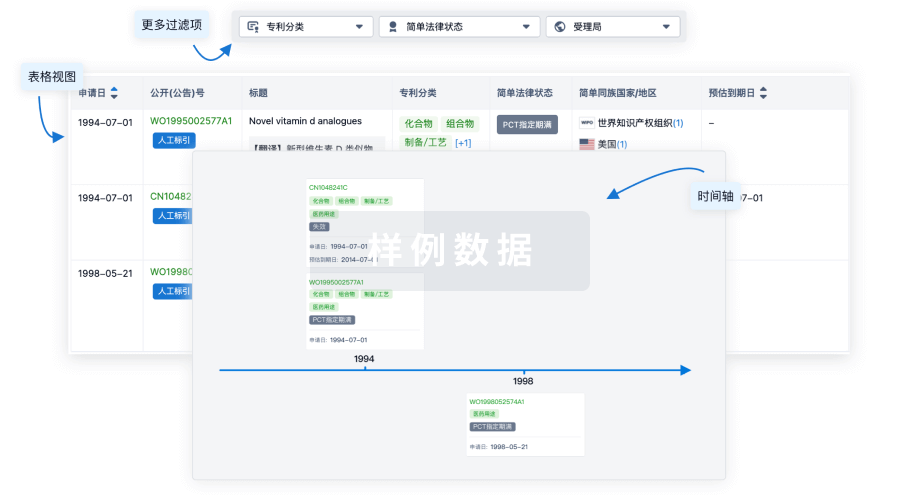
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
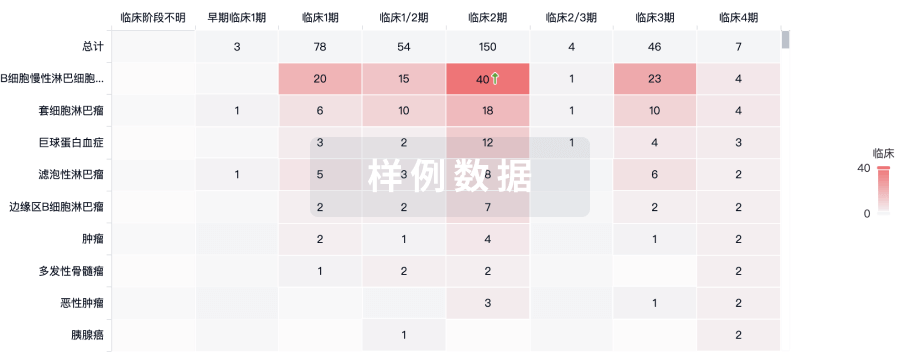
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用