预约演示
更新于:2025-03-20
Azurin-p28
更新于:2025-03-20
概要
基本信息
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评孤儿药 (美国) |




登录后查看时间轴
结构/序列
分子式C122H197N31O47S2 |
InChIKeyRFGPECAMUVWTJN-AWHPYGDNSA-N |
CAS号897026-25-4 |
Sequence Code 165904

来源: *****
关联
2
项与 Azurin-p28 相关的临床试验NCT01975116
A Phase I Trial of p28 (NSC745104), a Non-HDM2 Mediated Peptide Inhibitor of p53 Ubiquitination in Pediatric Patients With Recurrent or Progressive CNS Tumors
NCT00914914
A Phase I Trial of p28 (Cell Penetrating Peptide) in the Treatment of Refractory Solid Tumors
100 项与 Azurin-p28 相关的临床结果
登录后查看更多信息
100 项与 Azurin-p28 相关的转化医学
登录后查看更多信息
100 项与 Azurin-p28 相关的专利(医药)
登录后查看更多信息
44
项与 Azurin-p28 相关的文献(医药)2024-09-02Toxicology Research
Design and computational analysis of a novel Leptulipin-p28 fusion protein as a multitarget anticancer therapy in breast cancer
Article
作者: Nadeem, Tariq ; Bashir, Hamid ; Rehman, Hafiz Muhammad ; Khalid, Sania ; Al-Qassab, Yasamin ; Fatima, Tehreem ; Mubasher, Mian Muhammad ; Ahmad, Irfan ; Kalsoom, Maria
2023-10-13ACS Pharmacology & Translational Science
Novel Inhibitor of Mixed-Lineage Kinase Domain-Like Protein: The Antifibrotic Effects of a Necroptosis Antagonist
Article
作者: Bae, Sang Hyun ; Kwon, Ye-Mi ; Park, Sunyou ; Jun, Dae Won ; Lee, Jung Yeol ; Choi, Myeong A. ; Kim, Hye Young ; Hong, Eunmi ; Lee, Seung Min ; Lee, Ju Yeon ; Lee, A. Hyeon ; Park, Gye Ryeol ; Lee, Ji Hoon ; Oh, Ju Hee ; Park, Jin-wan
2023-07-01Molecular Cell
Glucose-induced CRL4COP1-p53 axis amplifies glycometabolism to drive tumorigenesis
Article
作者: Chen, Xing ; Huang, Niu ; Wang, Fengchao ; Zhang, Xiaozhe ; Zhao, Li ; Chen, Hao ; Luo, Yifan ; Su, Yang ; Xiao, Yihang ; Chow, Billy Kwok Chong ; Rao, Feng ; Wu, Mingxuan ; Wei, Xiayun ; Jin, Wenfei ; Zhang, Hongyun ; Fu, Qiuyu ; Nie, Siyue ; Zhang, Peitao ; Ren, Yan ; Wang, Huifang ; Pu, Weijie ; Hao, Yi ; Lin, Hong ; Zhang, Keren
100 项与 Azurin-p28 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 脉络丛肿瘤 | 药物发现 | 美国 | 2013-08-01 | |
| 神经胶质肉瘤 | 药物发现 | 美国 | 2013-08-01 | |
| 髓母细胞瘤 | 药物发现 | 美国 | 2013-08-01 | |
| 少突神经胶质瘤 | 药物发现 | 美国 | 2013-08-01 | |
| 实体瘤 | 药物发现 | 美国 | 2009-04-01 | |
| 实体瘤 | 药物发现 | 美国 | 2009-04-01 | |
| 脑癌 | 药物发现 | - | - | |
| 脑癌 | 药物发现 | - | - | |
| 脑癌 | 药物发现 | - | - | |
| 中枢神经系统肿瘤 | 药物发现 | 美国 | - | - |
登录后查看更多信息
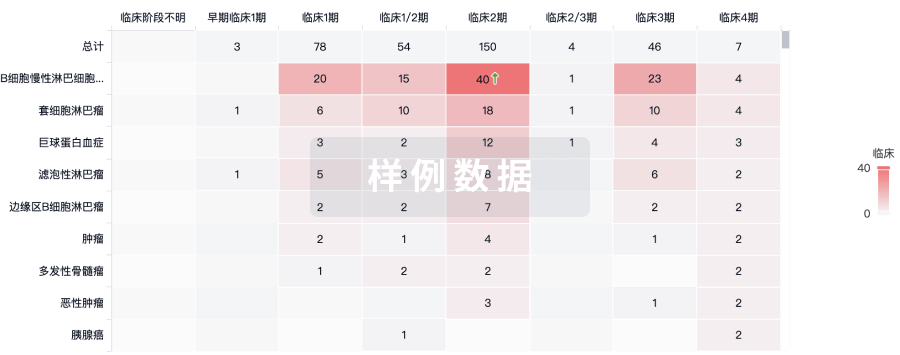
临床结果
临床结果
适应症
分期
评价
查看全部结果
登录后查看更多信息
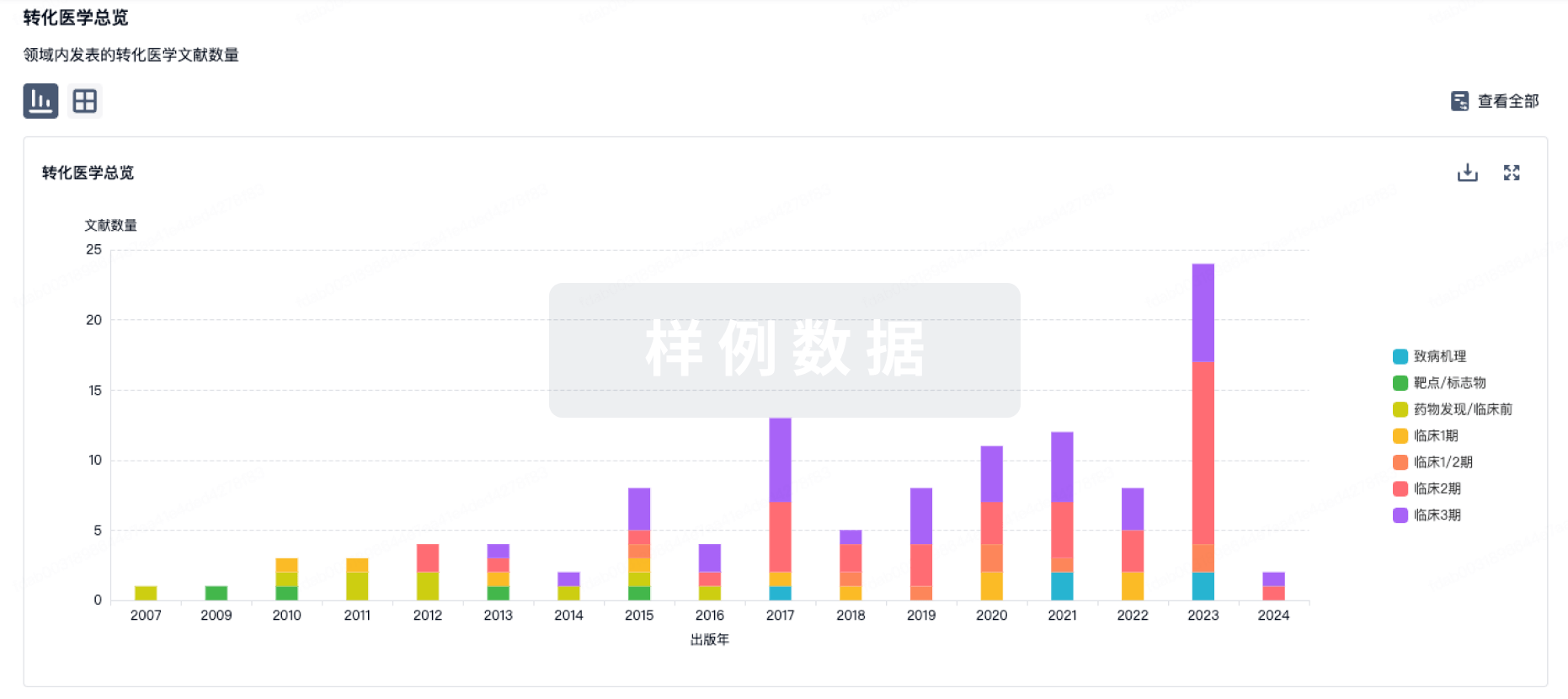
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
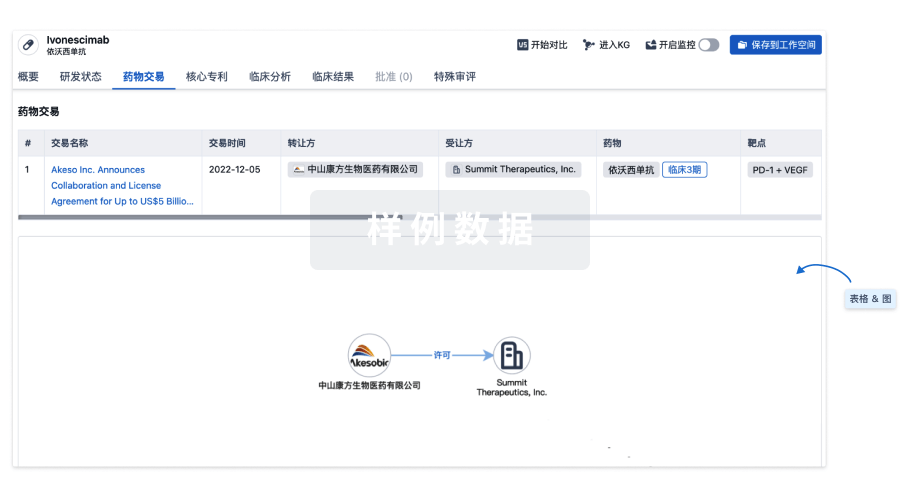
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
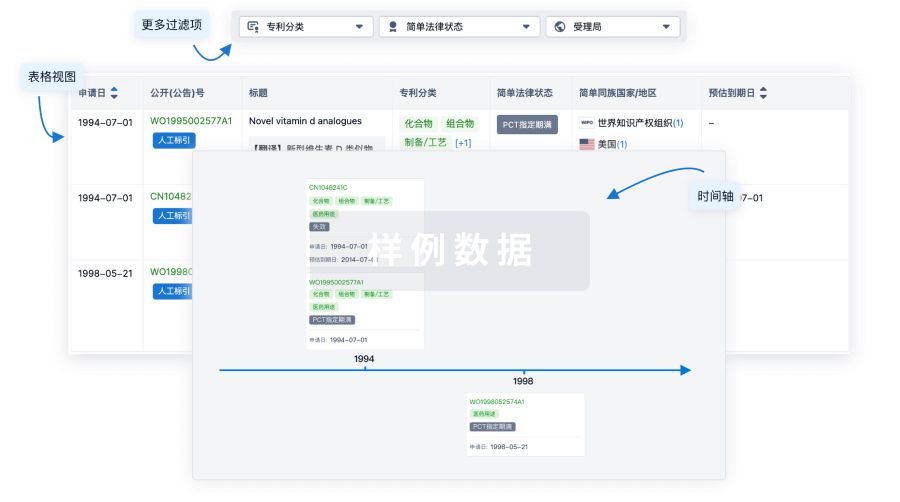
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
来和芽仔聊天吧
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用