预约演示
更新于:2025-05-07
CD30+ Anaplastic Large T-cell Lymphoma
CD30阳性间变性大T细胞淋巴瘤
更新于:2025-05-07
基本信息
别名- |
简介- |
关联
NCT02139592
Brentuximab Vedotin (ADCETRIS) IV Infusion - Special Drug Use Surveillance (All-case Surveillance) "Relapsed or Refractory CD30+ Hodgkin's Lymphoma or Anaplastic Large Cell Lymphoma"

100 项与 CD30阳性间变性大T细胞淋巴瘤 相关的临床结果
登录后查看更多信息
100 项与 CD30阳性间变性大T细胞淋巴瘤 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-04-17Blood
HSP-CAR30 with a high proportion of less-differentiated T cells promotes durable responses in refractory CD30+ lymphoma
Article
作者: Esquirol, Albert ; Moreno-Martinez, Maria-Estela ; Guardiola-Perello, Maria ; Riba, Mireia ; Garcia-Cadenas, Irene ; Jara-Bustamante, Paola ; Ujaldón-Miró, Cristina ; Alvarez-Fernández, Carmen ; Soria, José Manuel ; Escudero-López, Eva ; Escribà-Garcia, Laura ; Ezquerra, Pol ; Caballero, Ana Carolina ; Briones, Javier ; Sierra, Jorge ; Iranzo, Eva ; Montserrat-Torres, Rosanna ; Pujol-Fernández, Paula ; Martino, Rodrigo
2025-04-01The Lancet Haematology
Epidemiology, clinical features, and outcomes of peripheral T-cell lymphoma in Latin America: an international, retrospective, cohort study
Article
作者: Beltran, Brady ; Brasil, Sergio A B ; Pereyra, Patricio H ; Pereira, Juliana ; Federico, Massimo ; Roche, Claudia ; Pavlovsky, Astrid ; Fiad, Lorena ; Roa, Macarena ; Baptista, Renata L R ; Valvert, Fabiola ; Korin, Laura ; Malpica, Luis ; Vasquez, Jule F ; Mahuad, Carolina V ; Idrobo, Henry ; Oliver, Carolina ; Quiroz, Alfredo R ; Chiattone, Carlos ; Castro, Denisse ; Tavarez, Jamila Vaz ; Miranda, Eliana C M ; Warley, Fernando ; Duffles, Guilherme ; Gazitua, Raimundo ; Fischer, Thais ; Valcarcel, Bryan ; Torres, Maria A ; Villela, Luis ; Enriquez, Daniel J
2025-01-01Acta Pharmaceutica Sinica B
Dimeric natural product panepocyclinol A inhibits STAT3 via di-covalent modification
Article
作者: Zhou, Dawang ; Wang, Yiqiu ; Li, Yunzhan ; Deng, Qihong ; Wang, Yuezhou ; Li, Li ; Li, Xiaoyang ; Zhang, Lei ; Xu, Qingyan ; Yun, Cai-Hong ; Lian, Wenhua ; Zhang, Jianming ; Deng, Xianming ; Wei, Yanling ; Hu, Zhiyu ; Xia, Fei ; Chen, Lanfen ; Wu, Xiaobing ; Gui, Fu ; Gao, Fei ; Zhu, Su-Jie
分析
对领域进行一次全面的分析。
登录
或
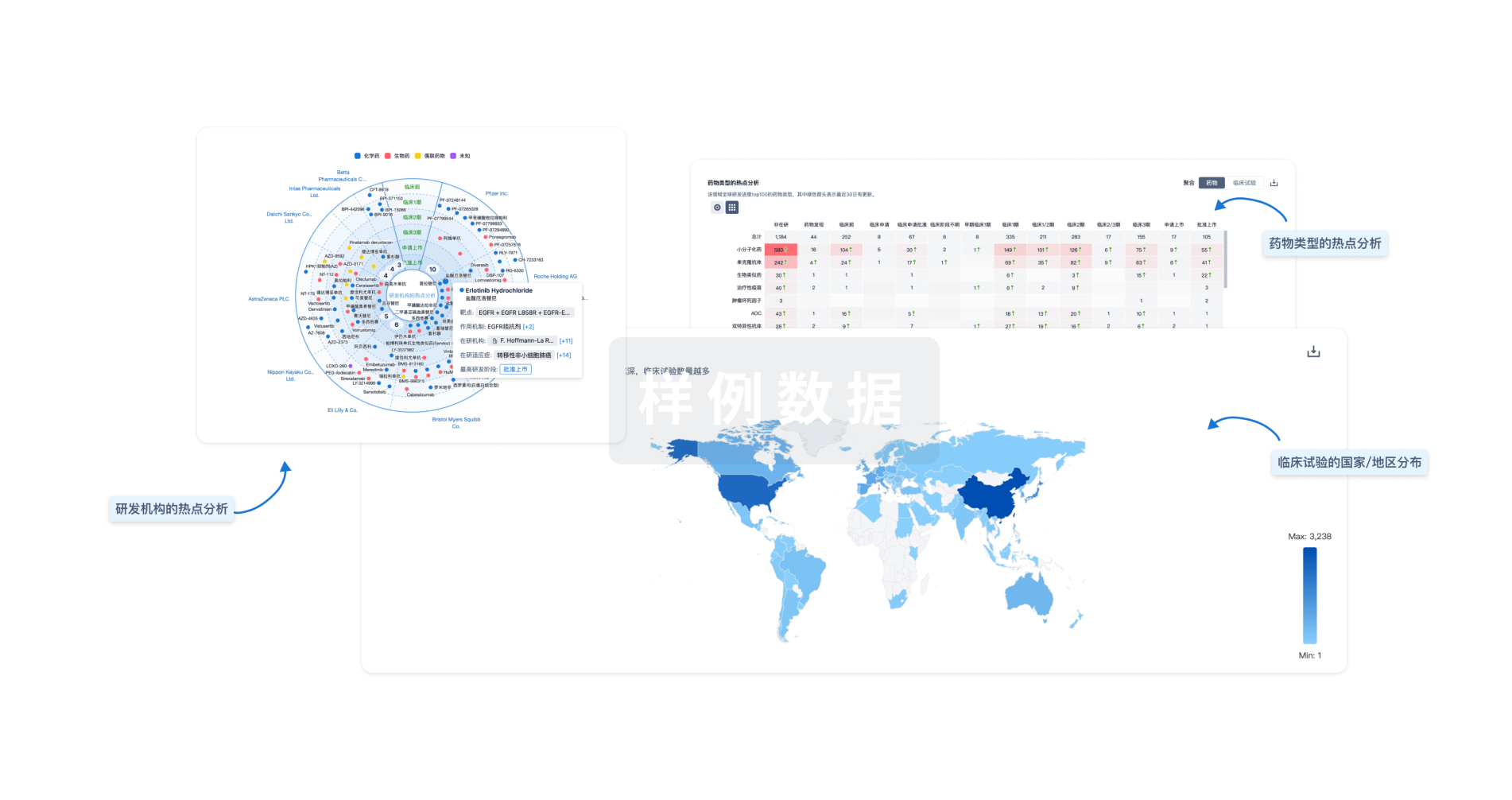
Eureka LS:
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用